

Медицинская генетика 2010 лекц 1.pptx
- Количество слайдов: 123
 Медицинская генетика кафедра факультетской педиатрии 2009
Медицинская генетика кафедра факультетской педиатрии 2009
 Система научных знаний, наблюдений за наследственностью и изменчивостью у человека на всех уровнях ее организации (молекулярном, клеточном, организменном, популяционном) называется клинической генетикой.
Система научных знаний, наблюдений за наследственностью и изменчивостью у человека на всех уровнях ее организации (молекулярном, клеточном, организменном, популяционном) называется клинической генетикой.
 АКСИОМЫ МЕДИЦИНСКОЙ ГЕНЕТИКИ ► Наследственные болезни являются частью общей наследственной изменчивости человека. ► В развитии наследственных признаков или болезней принимают участие наследственная конституция (генотип) и внешняя среда. Во всех жизненных проявлениях действие любых генов осуществляется в тесном взаимодействии с факторами среды
АКСИОМЫ МЕДИЦИНСКОЙ ГЕНЕТИКИ ► Наследственные болезни являются частью общей наследственной изменчивости человека. ► В развитии наследственных признаков или болезней принимают участие наследственная конституция (генотип) и внешняя среда. Во всех жизненных проявлениях действие любых генов осуществляется в тесном взаимодействии с факторами среды
 ► Человечество отягощено огромным «грузом» разнообразных мутаций, которые накапливались в процессе длительной эволюции. ► Наследственная отягощенность современного человечества состоит из двух компонент. Одна из них — накопленные в процессе эволюции и истории человечества патологические мутации, другая — вновь возникающие наследственные изменения в половых клетках. ► Человек постоянно сталкивается с новыми факторами среды, ранее никогда не встречавшимися на протяжении всей его эволюции, а также испытывает большие «нагрузки» социального и экологического характера. Это приводит к появлению новых видов наследственной патологии — экогенетических болезней.
► Человечество отягощено огромным «грузом» разнообразных мутаций, которые накапливались в процессе длительной эволюции. ► Наследственная отягощенность современного человечества состоит из двух компонент. Одна из них — накопленные в процессе эволюции и истории человечества патологические мутации, другая — вновь возникающие наследственные изменения в половых клетках. ► Человек постоянно сталкивается с новыми факторами среды, ранее никогда не встречавшимися на протяжении всей его эволюции, а также испытывает большие «нагрузки» социального и экологического характера. Это приводит к появлению новых видов наследственной патологии — экогенетических болезней.
 Тип патологии ► Генные болезни ► Распространёность, % Хромосомные болезни Болезни с существенным компонентом наследственной предрасположенности ► Генетические соматические нарушения ► Несовместимость матери и плода ► I (среди новорождённых) ► 0, 5 (среди новорождённых) ► ► 3— 3, 5 (среди детей до 5 лет) Неизвестна ► 0, 4 (среди новорождённых ►
Тип патологии ► Генные болезни ► Распространёность, % Хромосомные болезни Болезни с существенным компонентом наследственной предрасположенности ► Генетические соматические нарушения ► Несовместимость матери и плода ► I (среди новорождённых) ► 0, 5 (среди новорождённых) ► ► 3— 3, 5 (среди детей до 5 лет) Неизвестна ► 0, 4 (среди новорождённых ►
 ► Фенотип - индивидуальное развитие человека, формирование его внешних признаков. ► Фенотип есть реализация в конкретной внешней среде генотипа человека ► Генотип заложен в оплодотворенной яйцеклетке - зиготе ► Материальным носителем наследственной информации, передаваемой из поколения в поколение, есть хромосомы клеточных ядер. ► Хромосомы представлены двумя видами – аутосомами и половыми хромосомами. ► В организме есть 2 типа клеток – соматические и половые
► Фенотип - индивидуальное развитие человека, формирование его внешних признаков. ► Фенотип есть реализация в конкретной внешней среде генотипа человека ► Генотип заложен в оплодотворенной яйцеклетке - зиготе ► Материальным носителем наследственной информации, передаваемой из поколения в поколение, есть хромосомы клеточных ядер. ► Хромосомы представлены двумя видами – аутосомами и половыми хромосомами. ► В организме есть 2 типа клеток – соматические и половые
 Соматические клетки содержат полный набор 23 пары гомологичных хромосом (22 пары аутосом и 1 пара половых хромосом). ► Размножение соматических клеток – митоз ► В половых клетках содержится гаплоидный набор хромосом 23 хромосомы. ► Процесс деления половых клеток – мейоз. ► ► Нарушения в процессе мейоза и последующего митоза служит источником хромосомных болезней. ► В хромосомах в линейной последовательности в определенных местах (локусах) находятся дискретные единицы наследственности – гены.
Соматические клетки содержат полный набор 23 пары гомологичных хромосом (22 пары аутосом и 1 пара половых хромосом). ► Размножение соматических клеток – митоз ► В половых клетках содержится гаплоидный набор хромосом 23 хромосомы. ► Процесс деления половых клеток – мейоз. ► ► Нарушения в процессе мейоза и последующего митоза служит источником хромосомных болезней. ► В хромосомах в линейной последовательности в определенных местах (локусах) находятся дискретные единицы наследственности – гены.
 ► Идентичные локусы хромосом в гомологичных хромосомах называют аллелями. Идентичные аллели гомозиготны. Различные по структуре аллели - гетерозиготны. ►В гетерозиготном состоянии один из аллелей проявляется фенотипически – доминирует. Непроявляющийся аллель – рецессивен. ► Доминантными и рецессивными гены определяют как нормальные, так и патологические признаки
► Идентичные локусы хромосом в гомологичных хромосомах называют аллелями. Идентичные аллели гомозиготны. Различные по структуре аллели - гетерозиготны. ►В гетерозиготном состоянии один из аллелей проявляется фенотипически – доминирует. Непроявляющийся аллель – рецессивен. ► Доминантными и рецессивными гены определяют как нормальные, так и патологические признаки
 РОЛЬ НАСЛЕДСТВЕННОСТИ И СРЕДЫ В РАЗВИТИИ ПАТОЛОГИИ Любые проявления жизнедеятельности организма являются результатом взаимодействия наследственных и средовых факторов. Болезнь также развивается на основе тесного взаимодействия внешних повреждающих и внутренних факторов. ► С генетической точки зрения все болезни в зависимости от относительной значимости наследственных и средовых факторов в их развитии можно подразделить на 3 группы: наследственные болезни, болезни с наследственной предрасположенностью, ненаследственные болезни ► Наследственными болезнями называют такие болезни, ► этиологическим фактором которых являются мутации.
РОЛЬ НАСЛЕДСТВЕННОСТИ И СРЕДЫ В РАЗВИТИИ ПАТОЛОГИИ Любые проявления жизнедеятельности организма являются результатом взаимодействия наследственных и средовых факторов. Болезнь также развивается на основе тесного взаимодействия внешних повреждающих и внутренних факторов. ► С генетической точки зрения все болезни в зависимости от относительной значимости наследственных и средовых факторов в их развитии можно подразделить на 3 группы: наследственные болезни, болезни с наследственной предрасположенностью, ненаследственные болезни ► Наследственными болезнями называют такие болезни, ► этиологическим фактором которых являются мутации.
 ► О болезнях с наследственной предрасположенностью говорят тогда, когда болезнь развивается у лиц с определённой генетической характеристикой под влиянием факторов окружающей среды. ► Эти болезни называют также мультифакториальными. Наследственность — этиологический и патогенетический фактор. Для пенетрантности мутантных генов необходим соответствующий фактор окружающей среды. ► В происхождении ненаследственных болезней определяющую роль играет среда. Сюда относится большинство травм, инфекционных болезней, ожоги и т. д. Генетические факторы могут влиять только на течение патологических процессов (выздоровление, восстановительные процессы, компенсация нарушенных функций).
► О болезнях с наследственной предрасположенностью говорят тогда, когда болезнь развивается у лиц с определённой генетической характеристикой под влиянием факторов окружающей среды. ► Эти болезни называют также мультифакториальными. Наследственность — этиологический и патогенетический фактор. Для пенетрантности мутантных генов необходим соответствующий фактор окружающей среды. ► В происхождении ненаследственных болезней определяющую роль играет среда. Сюда относится большинство травм, инфекционных болезней, ожоги и т. д. Генетические факторы могут влиять только на течение патологических процессов (выздоровление, восстановительные процессы, компенсация нарушенных функций).
 Врожденные заболевания – состояния, существующие при рождении ребенка. Врожденные заболевания могут быть как наследственные, так и вызванные другими ненаследственными факторами. Ребенок с гидроцефалией, внутриутробно перенес токсоплазмоз – клинические проявления - поражение головного мозга, глаз, внутренних органов.
Врожденные заболевания – состояния, существующие при рождении ребенка. Врожденные заболевания могут быть как наследственные, так и вызванные другими ненаследственными факторами. Ребенок с гидроцефалией, внутриутробно перенес токсоплазмоз – клинические проявления - поражение головного мозга, глаз, внутренних органов.
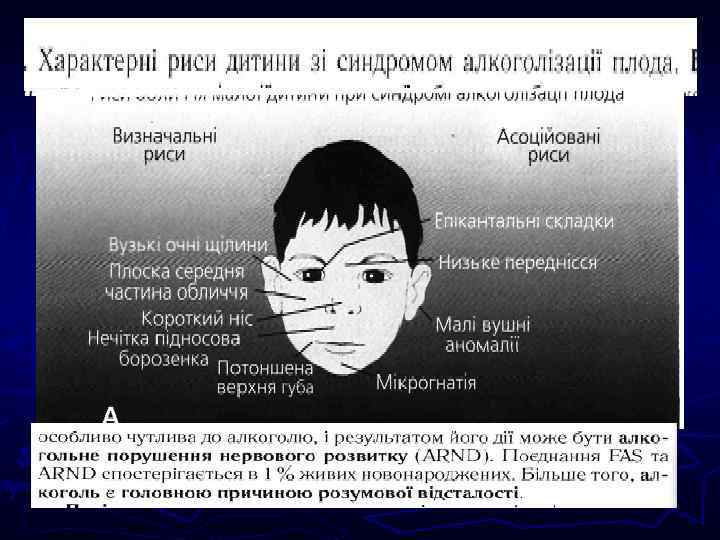
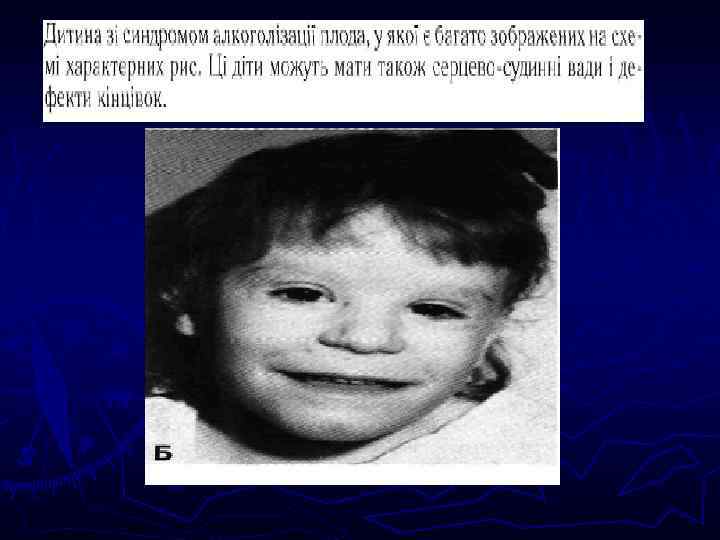
") Наследственные заболевания связаны с наследственными структурами организма. Подразделяются на заболевания генные (изменение структуры хромосом) и хромосомные (число хромосом). Типы хромосомных мутаций: числовые и структурные. Возможны в половых и соматических клетках. ► Числовые мутации - добавочные хромосомы (трисомии) или отсутствие одной или части хромосом (моносомии). Либо увеличение на полный гаплоидный набор хромосом (три, тетраплоидии) ► Структурные изменения – делеция –утрата части хромосомы из-за 2 -х разрывов и одного воссоединения Х с утратой сегмента, дупликация – удвоение сегмента хромосомы (полиплоидность по данному сегменту), инверсия – поворот участка Х на 180 гр. и воссоединение разорванных концов в новом порядке, транслокация – обмен сегментов между хромосомами ►
Наследственные заболевания связаны с наследственными структурами организма. Подразделяются на заболевания генные (изменение структуры хромосом) и хромосомные (число хромосом). Типы хромосомных мутаций: числовые и структурные. Возможны в половых и соматических клетках. ► Числовые мутации - добавочные хромосомы (трисомии) или отсутствие одной или части хромосом (моносомии). Либо увеличение на полный гаплоидный набор хромосом (три, тетраплоидии) ► Структурные изменения – делеция –утрата части хромосомы из-за 2 -х разрывов и одного воссоединения Х с утратой сегмента, дупликация – удвоение сегмента хромосомы (полиплоидность по данному сегменту), инверсия – поворот участка Х на 180 гр. и воссоединение разорванных концов в новом порядке, транслокация – обмен сегментов между хромосомами ►
 Этиологический, т. е. характеристика хромосомной или") КЛАССИФИКАЦИЯ хромосомных заболеваний основана на нескольких принципах a) Этиологический, т. е. характеристика хромосомной или геномной мутации: I. Хромосомные болезни, связанные с аномалиями числа хромосом при сохранении их структуры. 1) II. Хромосомные болезни, обусловленные структурными перестройками хромосом. Виды хромосомных аберраций: 1).
КЛАССИФИКАЦИЯ хромосомных заболеваний основана на нескольких принципах a) Этиологический, т. е. характеристика хромосомной или геномной мутации: I. Хромосомные болезни, связанные с аномалиями числа хромосом при сохранении их структуры. 1) II. Хромосомные болезни, обусловленные структурными перестройками хромосом. Виды хромосомных аберраций: 1).
: ► Гаметические мутации") Определение типа клеток, в которых возникла мутация (в гаметах или зиготе): ► Гаметические мутации ведут к полным формам хромосомных болезней. У таких индивидов все клетки несут унаследованную с гаметой хромосомную аномалию. ► Соматические мутации - если аномалия возникает в зиготе или на ранних стадиях дробления, при этом развивается организм с клетками разной хромосомной конституции (два типа и более). Это явление называется мозаицизм, а формы хромосомных болезней мозаичными. Для того, чтобы мозаичная форма по клинической картине совпадала с полной, необходимо иметь не менее 10% клеток с аномальным набором. c) Время возникновения мутации (в поколении): ► - Спорадические случаи - мутация возникла заново в гаметах здоровых родителей или на стадии зиготы. ► - Наследуемые (семейные) формы - когда родители уже имели подобную аномалию.
Определение типа клеток, в которых возникла мутация (в гаметах или зиготе): ► Гаметические мутации ведут к полным формам хромосомных болезней. У таких индивидов все клетки несут унаследованную с гаметой хромосомную аномалию. ► Соматические мутации - если аномалия возникает в зиготе или на ранних стадиях дробления, при этом развивается организм с клетками разной хромосомной конституции (два типа и более). Это явление называется мозаицизм, а формы хромосомных болезней мозаичными. Для того, чтобы мозаичная форма по клинической картине совпадала с полной, необходимо иметь не менее 10% клеток с аномальным набором. c) Время возникновения мутации (в поколении): ► - Спорадические случаи - мутация возникла заново в гаметах здоровых родителей или на стадии зиготы. ► - Наследуемые (семейные) формы - когда родители уже имели подобную аномалию.
 Транслокации - перенос участка из одной хромосомы в другую") Хромосомные аберрации (структурные нарушения ) Транслокации - перенос участка из одной хромосомы в другую или в другое место этой же хромосомы. Исходы - гибель, ВПР, высокий риск рождения больных детей. Например, слияние 2 -х хромосом в одну (синдром Дауна) - 21 хромосома с 14 -ой или 15 -ой. ► Инверсия - при разрыве хромосомы в двух местах освобожденный участок разворачивается на 180% и вновь встает на прежнее место. Исходы - спонтанные аборты, множественные ВПР, малые аномалии развития, умственная отсталость, без аномалий. ► Делеция - исчезновение оторванной части хромосом. ►
Хромосомные аберрации (структурные нарушения ) Транслокации - перенос участка из одной хромосомы в другую или в другое место этой же хромосомы. Исходы - гибель, ВПР, высокий риск рождения больных детей. Например, слияние 2 -х хромосом в одну (синдром Дауна) - 21 хромосома с 14 -ой или 15 -ой. ► Инверсия - при разрыве хромосомы в двух местах освобожденный участок разворачивается на 180% и вновь встает на прежнее место. Исходы - спонтанные аборты, множественные ВПР, малые аномалии развития, умственная отсталость, без аномалий. ► Делеция - исчезновение оторванной части хромосом. ►
 ► Структурные изменения хромосом: § делеция – утрата части хромосомы изза 2 -х разрывов и одного воссоединения хромосомы с утратой сегмента, § дупликация – удвоение сегмента хромосомы (полиплоидность по данному сегменту), § инверсия – поворот участка хромосомы на 180 гр. и воссоединение разорванных концов в новом порядке, § транслокация – обмен сегментов между хромосомами
► Структурные изменения хромосом: § делеция – утрата части хромосомы изза 2 -х разрывов и одного воссоединения хромосомы с утратой сегмента, § дупликация – удвоение сегмента хромосомы (полиплоидность по данному сегменту), § инверсия – поворот участка хромосомы на 180 гр. и воссоединение разорванных концов в новом порядке, § транслокация – обмен сегментов между хромосомами
. Генные мутации наследуются в строгом") Генные – болезни, вызванные генными мутациями. (изменение структуры ДНК). Генные мутации наследуются в строгом соответствии с законами Менделя в зависимости от рецессивности или доминантности генов. ► Существуют доминантный и рецессивный тип наследования заболеваний. При доминантном типе наследования от больного родителя сибс получает хотя бы один патологический ген. При рецессивном типе наследования – от обоих родителей 2 идентичных патологических аллеля. ► С учетом локализации генов и характеристики их доминантности наследование бывает: § Аутосомно –доминантное § аутосомно –рецессивное § доминантное, сцепленное с х –хромосомой § рецессивное, сцепленное с х – хромосомой
Генные – болезни, вызванные генными мутациями. (изменение структуры ДНК). Генные мутации наследуются в строгом соответствии с законами Менделя в зависимости от рецессивности или доминантности генов. ► Существуют доминантный и рецессивный тип наследования заболеваний. При доминантном типе наследования от больного родителя сибс получает хотя бы один патологический ген. При рецессивном типе наследования – от обоих родителей 2 идентичных патологических аллеля. ► С учетом локализации генов и характеристики их доминантности наследование бывает: § Аутосомно –доминантное § аутосомно –рецессивное § доминантное, сцепленное с х –хромосомой § рецессивное, сцепленное с х – хромосомой
 Клиническая картина генных заболеваний определяется пенетрантностью и вариациями экспрессии мутации ► Экспрессивность – степень проявления признака, соответствует тяжести течения в клиническом понимании. (нейрофиброматоз) ► Пенетрантность – это частота или вероятность проявления. Бывает полной и неполной. Полная пенетрантность - ген проявляется у каждого человека. Если в некоторых семьях его фенотипическое проявление выпадает, то говорят о неполной пенетрантности. (недостаточность глюкозо - 6 – фосфатдегидрогеназы).
Клиническая картина генных заболеваний определяется пенетрантностью и вариациями экспрессии мутации ► Экспрессивность – степень проявления признака, соответствует тяжести течения в клиническом понимании. (нейрофиброматоз) ► Пенетрантность – это частота или вероятность проявления. Бывает полной и неполной. Полная пенетрантность - ген проявляется у каждого человека. Если в некоторых семьях его фенотипическое проявление выпадает, то говорят о неполной пенетрантности. (недостаточность глюкозо - 6 – фосфатдегидрогеназы).

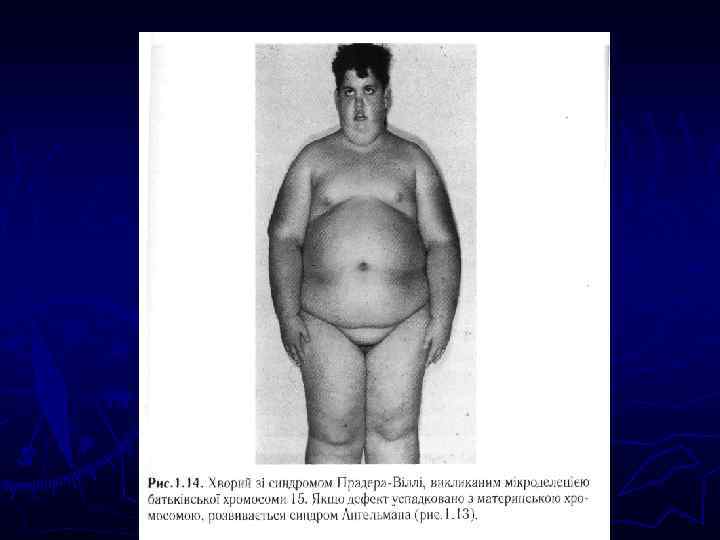
 Понятие геномного импринтинга – это понятие «новой генетики» - функциональной моносомии. ► В геноме есть две хромосомы, но независимо от доминантности или рецесивности признака может работать только одна из них. ► Синдром Прадера Вилли – мужское проявление и с-м Ангельмана – материнского происхождения. В первом случае в 15 хромосоме, в ее коротком плече наблюдается проявление отцовского гена, материнский заблокирован. Во втором случае в той же хромосоме, в том же локусе блокирован мужской ген.
Понятие геномного импринтинга – это понятие «новой генетики» - функциональной моносомии. ► В геноме есть две хромосомы, но независимо от доминантности или рецесивности признака может работать только одна из них. ► Синдром Прадера Вилли – мужское проявление и с-м Ангельмана – материнского происхождения. В первом случае в 15 хромосоме, в ее коротком плече наблюдается проявление отцовского гена, материнский заблокирован. Во втором случае в той же хромосоме, в том же локусе блокирован мужской ген.
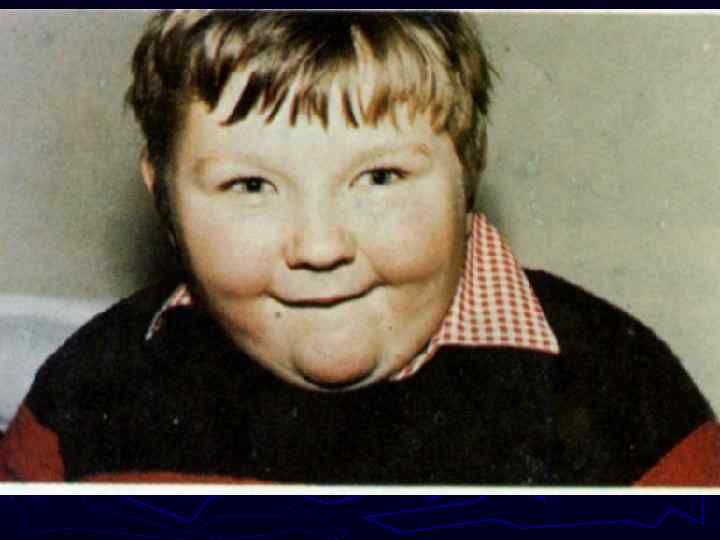

 Моногенные болезни ► составляют значительную долю наследственной патологии и насчитывают сегодня более 4500 заболеваний ► выявляются у 30 -65 детей на 1000 новорожденных, что составляет 3, 0 -6, 5%, ► в структуре общей смертности детей до 5 лет на их долю приходится 10 -14%. ► представляют значительные трудности в своевременной диагностике и эффективном лечении ► приводят к значительному нарушению качества жизни больных, инвалидизации и раннему летальному исходу.
Моногенные болезни ► составляют значительную долю наследственной патологии и насчитывают сегодня более 4500 заболеваний ► выявляются у 30 -65 детей на 1000 новорожденных, что составляет 3, 0 -6, 5%, ► в структуре общей смертности детей до 5 лет на их долю приходится 10 -14%. ► представляют значительные трудности в своевременной диагностике и эффективном лечении ► приводят к значительному нарушению качества жизни больных, инвалидизации и раннему летальному исходу.
 Заболевания многочислены и отличаются выраженным клиническим полиморфизмом. Классификация ► ► 1. По ведущей системной патологии - по органному и системному типу. 2. По этиологии. - болезни с установленным первичным молекулярным (биохимическим) дефектом. Число таких болезней продолжает неуклонно расти: если по каталогу Маккьюсика в 1990 г. этот класс составил около 100 болезней, то на конец 1993 г. их было уже 328, а к 2001 г. насчитывалось около 500 (10 -11% всех МБ); - болезни с неустановленным первичным молекулярным (биохимическим) дефектом. На эти заболевания приходится около 90% всех МБ. Первичный молекулярный дефект подразумевает определение дефектного гена и установления вида его конкретных изменений, появление и передача которых по наследству определяет развитие болезни, т. е. речь идет об изменениях в генетическом локусе. ► Первичный биохимический дефект подразумевает уровень простой биохимической реакции (функции), в осуществлении которой участвует белковый продукт нормального гена и которая первично нарушается из-за соответствующнго дефекта в структуре белка. ►
Заболевания многочислены и отличаются выраженным клиническим полиморфизмом. Классификация ► ► 1. По ведущей системной патологии - по органному и системному типу. 2. По этиологии. - болезни с установленным первичным молекулярным (биохимическим) дефектом. Число таких болезней продолжает неуклонно расти: если по каталогу Маккьюсика в 1990 г. этот класс составил около 100 болезней, то на конец 1993 г. их было уже 328, а к 2001 г. насчитывалось около 500 (10 -11% всех МБ); - болезни с неустановленным первичным молекулярным (биохимическим) дефектом. На эти заболевания приходится около 90% всех МБ. Первичный молекулярный дефект подразумевает определение дефектного гена и установления вида его конкретных изменений, появление и передача которых по наследству определяет развитие болезни, т. е. речь идет об изменениях в генетическом локусе. ► Первичный биохимический дефект подразумевает уровень простой биохимической реакции (функции), в осуществлении которой участвует белковый продукт нормального гена и которая первично нарушается из-за соответствующнго дефекта в структуре белка. ►
, - аутосомно-рецессивные (Р), - сцепленные") 3. По типу наследования патологического признака - аутосомно-доминантные (Д), - аутосомно-рецессивные (Р), - сцепленные с половой хромосомой (Х-сц) доминантные и рецессивные - митохондриальные. 4. По преимущественному поражению того или иного вида обмена. ► многие МБ называются наследственными болезнями обмена веществ (НБО). ► выделено более 700 форм, в том числе 200 с установленным биохимическим дефектом.
3. По типу наследования патологического признака - аутосомно-доминантные (Д), - аутосомно-рецессивные (Р), - сцепленные с половой хромосомой (Х-сц) доминантные и рецессивные - митохондриальные. 4. По преимущественному поражению того или иного вида обмена. ► многие МБ называются наследственными болезнями обмена веществ (НБО). ► выделено более 700 форм, в том числе 200 с установленным биохимическим дефектом.
;") ► ► ► 1 болезни аминокислотного обмена (ФКУ, тирозиноз, алкаптонурия, лейциноз и др. ); 2 болезни углеводного обмена (галактоземия, гликогенозы, мукополисахаридозы); 3 болезни липидного обмена (эссенциальные семейные липидозы, ганглиозидозы, сфинголипидозы, цереброзидозы, лейкодистрофии, гиперлипидемии и др. ); 4 болезни биосинтеза кортикостероидов (адрено-генитальный синдром, гипоальдостеронизм и др. ); 5 болезни пуринового и пиримидинового обмена (оротовая ацидурия, подагра и др. ); 6 болезни порфиринового и билирубинового обмена (синдромы Жильбера, Криглера-Найяра, порфирии и др. ); 7 болезни эритрона (анемия Фанкони, гемолитические анемии, дефицит глюкозо-6 -фосфатдегидрогеназы и др. ); 8 болезни металлов (болезни Вильсона-Коновалова, Менкеса, семейный пери-одический паралич и др. ); 9 болезни транспорта систем почек (болезнь де Тони-Дебре-Фанкони, витамин D-резистентный рахит, тубулопатии и др. ); 10 болезни лимфоцитов и лейкоцитов (недостаточность аденозиндезаминазы, септический гранулематоз и др. ). 11 болезни накопления (тезаурисмозы). Эта группа заболеваний, вызываемых недостатком лизосомальных ферментов и проявляющихся прогрессирующим отложением веществ определенного типа (обычно предшественники реакций) в клетках различных тканей - гликогенозы, цереброзидозы и др.
► ► ► 1 болезни аминокислотного обмена (ФКУ, тирозиноз, алкаптонурия, лейциноз и др. ); 2 болезни углеводного обмена (галактоземия, гликогенозы, мукополисахаридозы); 3 болезни липидного обмена (эссенциальные семейные липидозы, ганглиозидозы, сфинголипидозы, цереброзидозы, лейкодистрофии, гиперлипидемии и др. ); 4 болезни биосинтеза кортикостероидов (адрено-генитальный синдром, гипоальдостеронизм и др. ); 5 болезни пуринового и пиримидинового обмена (оротовая ацидурия, подагра и др. ); 6 болезни порфиринового и билирубинового обмена (синдромы Жильбера, Криглера-Найяра, порфирии и др. ); 7 болезни эритрона (анемия Фанкони, гемолитические анемии, дефицит глюкозо-6 -фосфатдегидрогеназы и др. ); 8 болезни металлов (болезни Вильсона-Коновалова, Менкеса, семейный пери-одический паралич и др. ); 9 болезни транспорта систем почек (болезнь де Тони-Дебре-Фанкони, витамин D-резистентный рахит, тубулопатии и др. ); 10 болезни лимфоцитов и лейкоцитов (недостаточность аденозиндезаминазы, септический гранулематоз и др. ). 11 болезни накопления (тезаурисмозы). Эта группа заболеваний, вызываемых недостатком лизосомальных ферментов и проявляющихся прогрессирующим отложением веществ определенного типа (обычно предшественники реакций) в клетках различных тканей - гликогенозы, цереброзидозы и др.
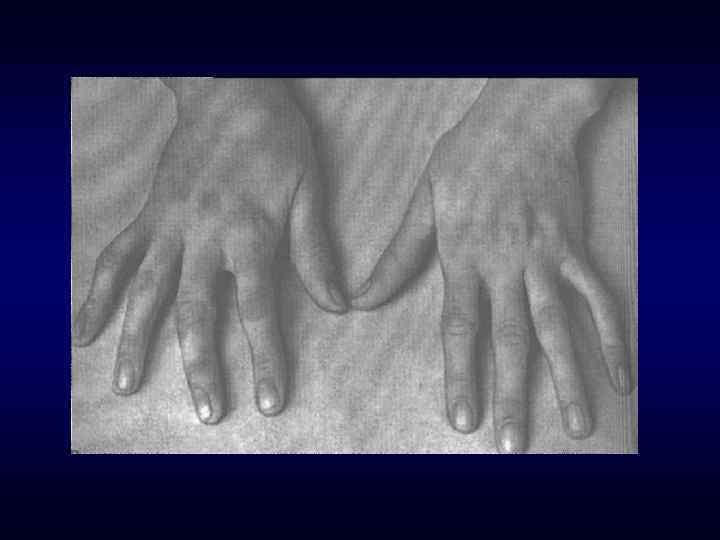
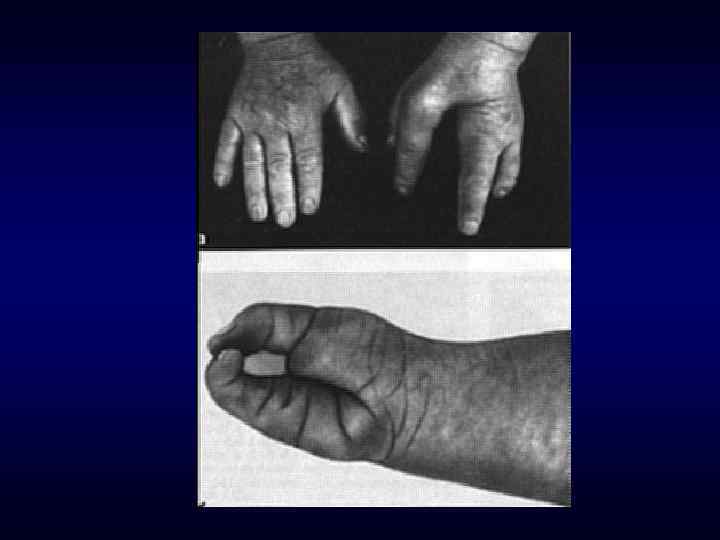
 Семейственность. Хроническое, прогредиентное, рецидивирующее течение. ► Наличие") Семиотика наследственных заболеваний (общие особенности клинических проявлений) Семейственность. Хроническое, прогредиентное, рецидивирующее течение. ► Наличие специфических симптомов или их сочетание. ► Вовлечение в патологический процесс первично многих органов или систем органов (свойство генов - эффект плейотропии). ► Резистентность к наиболее распространенным методам терапии. ► Фенотипические особенности индивида ► ► ► Антропометрия. ► Наличие врожденных пороков развития. ► Наличие микро аномалий развития / врожденных морфогенетических вариант.
Семиотика наследственных заболеваний (общие особенности клинических проявлений) Семейственность. Хроническое, прогредиентное, рецидивирующее течение. ► Наличие специфических симптомов или их сочетание. ► Вовлечение в патологический процесс первично многих органов или систем органов (свойство генов - эффект плейотропии). ► Резистентность к наиболее распространенным методам терапии. ► Фенотипические особенности индивида ► ► ► Антропометрия. ► Наличие врожденных пороков развития. ► Наличие микро аномалий развития / врожденных морфогенетических вариант.
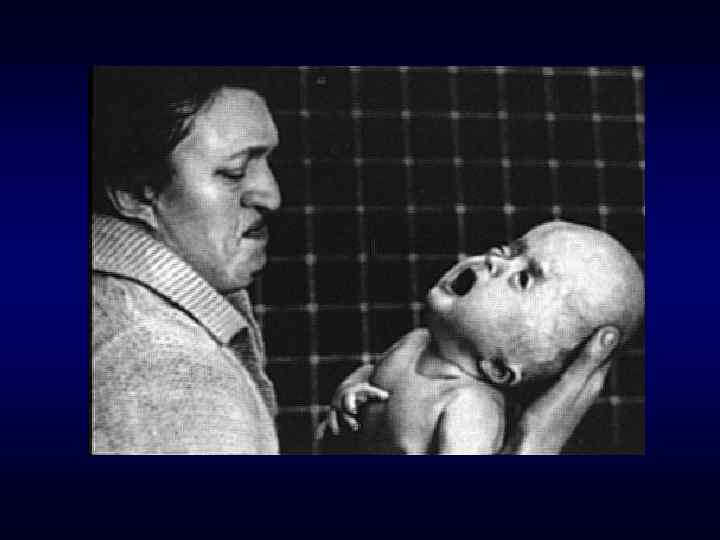
 Хроническое, прогредиентное, рецидивирующее течение. ► Наличие специфических") Семиотика наследственных заболеваний (общие особенности клинических проявлений) Хроническое, прогредиентное, рецидивирующее течение. ► Наличие специфических симптомов или их сочетание. ► Вовлечение в патологический процесс первично многих органов или систем органов (свойство генов - эффект плейотропии). ► Врожденный характер заболеваний ► Резистентность к наиболее распространенным методам терапии. ► Фенотипических особенности индивида ► ► Антропометрия. ► Наличие врожденных пороков развития. ► Наличие микро аномалий развития / врожденных морфогенетических вариант.
Семиотика наследственных заболеваний (общие особенности клинических проявлений) Хроническое, прогредиентное, рецидивирующее течение. ► Наличие специфических симптомов или их сочетание. ► Вовлечение в патологический процесс первично многих органов или систем органов (свойство генов - эффект плейотропии). ► Врожденный характер заболеваний ► Резистентность к наиболее распространенным методам терапии. ► Фенотипических особенности индивида ► ► Антропометрия. ► Наличие врожденных пороков развития. ► Наличие микро аномалий развития / врожденных морфогенетических вариант.
 ► Патологический фенотип определенного наследственного синдрома складывается из более или менее устойчивого сочетания отдельных симптомов (минимальные диагностические признаки), создающих в совокупности специфическое «фенотипическое ядро» заболевания, являющееся основой для установления диагноза. ► Синдром - совокупность внешних и внутренних, морфологических и функциональных аномалий и врожденных пороков, вызванных единым морфологическим фактором.
► Патологический фенотип определенного наследственного синдрома складывается из более или менее устойчивого сочетания отдельных симптомов (минимальные диагностические признаки), создающих в совокупности специфическое «фенотипическое ядро» заболевания, являющееся основой для установления диагноза. ► Синдром - совокупность внешних и внутренних, морфологических и функциональных аномалий и врожденных пороков, вызванных единым морфологическим фактором.
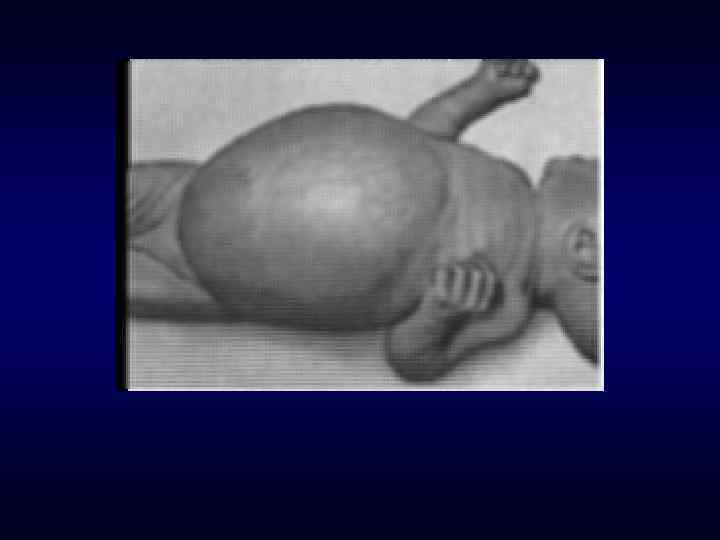
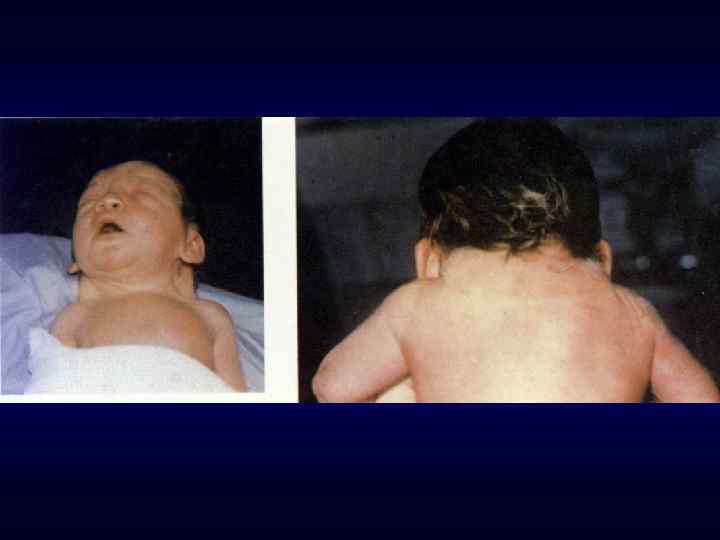
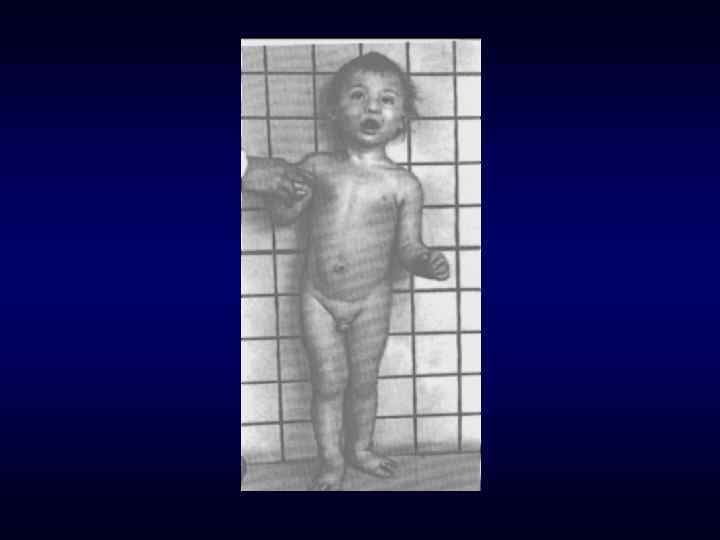
 Под термином «врождённый порок развития» понимают морфологический дефект органа, части органа или большой области тела, ведущий к нарушению функции органа(ов). ► Врожденные пороки могут быть изолированные, системные и множественные (в органах 2 и более систем). В 30 % СМПР является хромосомным заболеванием. ► Наследственно обусловленные врождённые пороки развития возникают у индивидов при наличии либо генных мутаций, эффект которых проявляется ввиде эмбрионального дисморфогенеза, либо хромосомных и геномных мутаций (хромосомные болезни). ►
Под термином «врождённый порок развития» понимают морфологический дефект органа, части органа или большой области тела, ведущий к нарушению функции органа(ов). ► Врожденные пороки могут быть изолированные, системные и множественные (в органах 2 и более систем). В 30 % СМПР является хромосомным заболеванием. ► Наследственно обусловленные врождённые пороки развития возникают у индивидов при наличии либо генных мутаций, эффект которых проявляется ввиде эмбрионального дисморфогенеза, либо хромосомных и геномных мутаций (хромосомные болезни). ►
 Экзогенно обусловленные пороки развития являются следствием действия тератогенных факторов в эмбриональном периоде, когда осуществляется органогенез. Механизм их действия во многом неясен. Тератогены могут оказывать цитоповреждающее действие, вызывать нарушение дифференцировки клеток в зачатках органов или мутации. ► Хорошо доказано тератогенное действие ионизирующей радиации, лекарственных веществ (талидомид, диазепам, гидантоин, варфарин, вальпроевая кислота, аминоптерин, стероидные гормоны и др. ), вредных привычек (курение, алкоголь), недостаточного питания (дефицит витаминов и микроэлементов), биологических факторов (краснуха, цитомегалия). ►
Экзогенно обусловленные пороки развития являются следствием действия тератогенных факторов в эмбриональном периоде, когда осуществляется органогенез. Механизм их действия во многом неясен. Тератогены могут оказывать цитоповреждающее действие, вызывать нарушение дифференцировки клеток в зачатках органов или мутации. ► Хорошо доказано тератогенное действие ионизирующей радиации, лекарственных веществ (талидомид, диазепам, гидантоин, варфарин, вальпроевая кислота, аминоптерин, стероидные гормоны и др. ), вредных привычек (курение, алкоголь), недостаточного питания (дефицит витаминов и микроэлементов), биологических факторов (краснуха, цитомегалия). ►
 Наличие специфических симптомов или их сочетание. ►") Семиотика наследственных заболеваний (общие особенности клинических проявлений) Наличие специфических симптомов или их сочетание. ► Вовлечение в патологический процесс первично многих органов или систем органов (свойство генов - эффект плейотропии). ► Врожденный характер заболеваний ► Резистентность к наиболее распространенным методам терапии. ► Фенотипических особенности индивида ► ► Антропометрия. ► Наличие врожденных пороков развития. ► Наличие микро аномалий развития / врожденных морфогенетических вариант.
Семиотика наследственных заболеваний (общие особенности клинических проявлений) Наличие специфических симптомов или их сочетание. ► Вовлечение в патологический процесс первично многих органов или систем органов (свойство генов - эффект плейотропии). ► Врожденный характер заболеваний ► Резистентность к наиболее распространенным методам терапии. ► Фенотипических особенности индивида ► ► Антропометрия. ► Наличие врожденных пороков развития. ► Наличие микро аномалий развития / врожденных морфогенетических вариант.
 составляют") ► из общего количества врождённых пороков развития генетически обусловленные формы (генные и хромосомные) составляют примерно 20— 30%, мультифакториальные — 30— 40%, экзогенные (тератогенные) — 2— 5%, неясной этиологии — 25— 50%. ► В зависимости от стадии онтогенеза, когда патогенный фактор действовал на развитие организма, врождённые пороки развития бывают следствием гаметопатий, бластопатий, эмбриопатий и фетопатий.
► из общего количества врождённых пороков развития генетически обусловленные формы (генные и хромосомные) составляют примерно 20— 30%, мультифакториальные — 30— 40%, экзогенные (тератогенные) — 2— 5%, неясной этиологии — 25— 50%. ► В зависимости от стадии онтогенеза, когда патогенный фактор действовал на развитие организма, врождённые пороки развития бывают следствием гаметопатий, бластопатий, эмбриопатий и фетопатий.
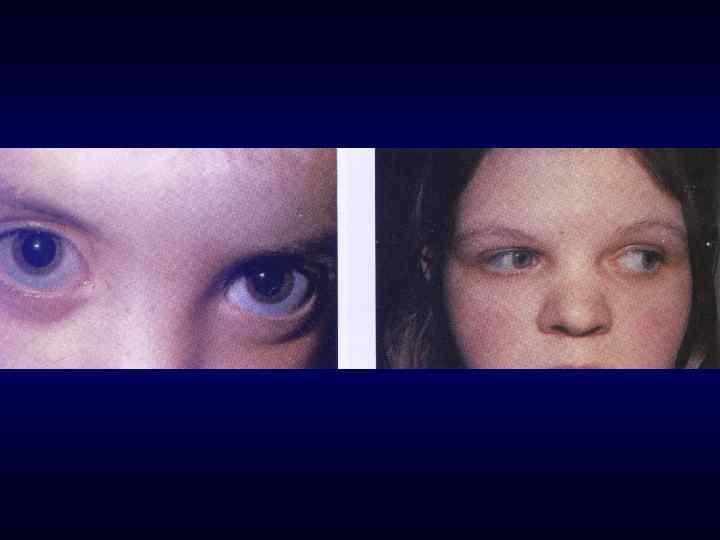
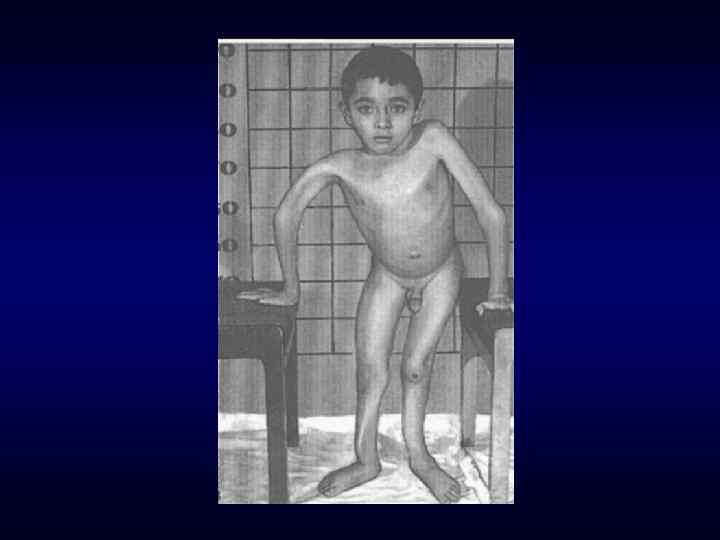
 ► Вовлечение в патологический процесс первично многих") Семиотика наследственных заболеваний (общие особенности клинических проявлений) ► Вовлечение в патологический процесс первично многих органов или систем органов (свойство генов - эффект плейотропии). ► Врожденный характер заболеваний ► Резистентность к наиболее распространенным методам терапии. ► Фенотипических особенности индивида ►Антропометрия. ►Наличие врожденных пороков развития. ►Наличие микро аномалий развития / врожденных морфогенетических вариант.
Семиотика наследственных заболеваний (общие особенности клинических проявлений) ► Вовлечение в патологический процесс первично многих органов или систем органов (свойство генов - эффект плейотропии). ► Врожденный характер заболеваний ► Резистентность к наиболее распространенным методам терапии. ► Фенотипических особенности индивида ►Антропометрия. ►Наличие врожденных пороков развития. ►Наличие микро аномалий развития / врожденных морфогенетических вариант.
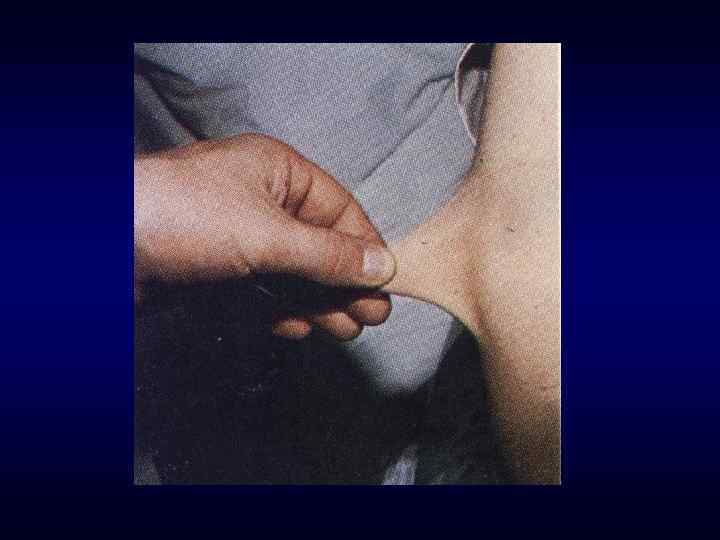
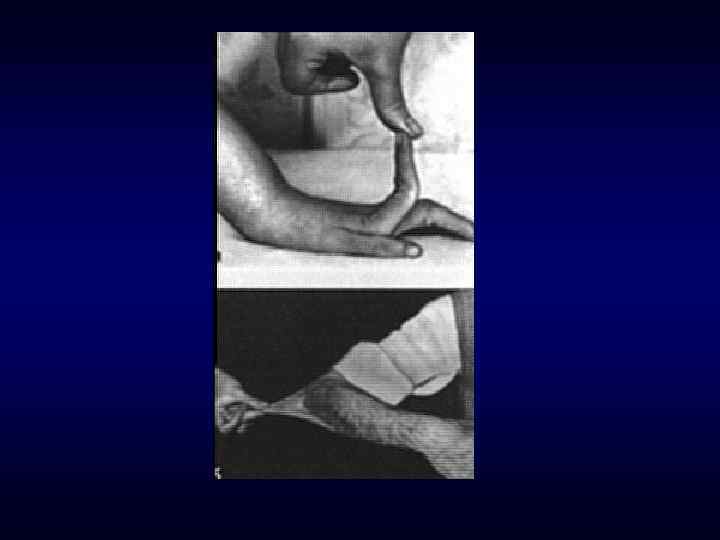

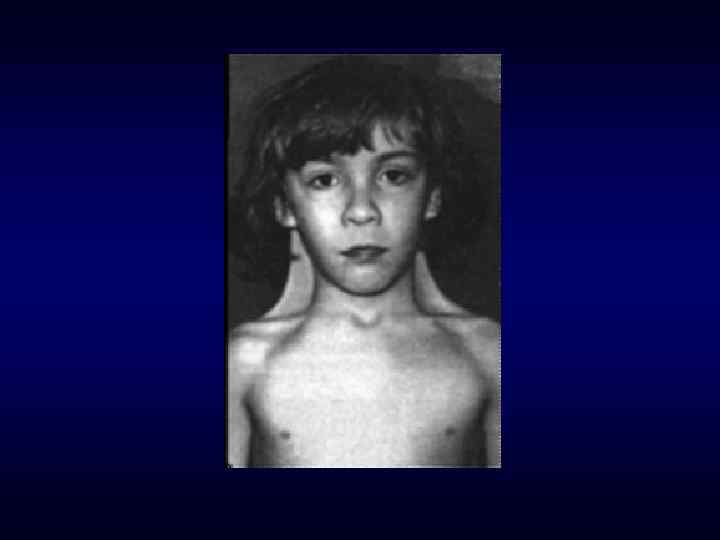
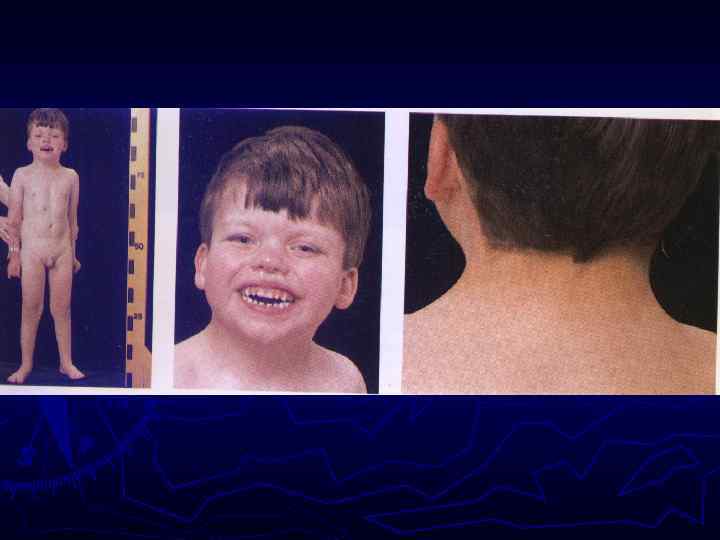
 Синдром Марфана
Синдром Марфана
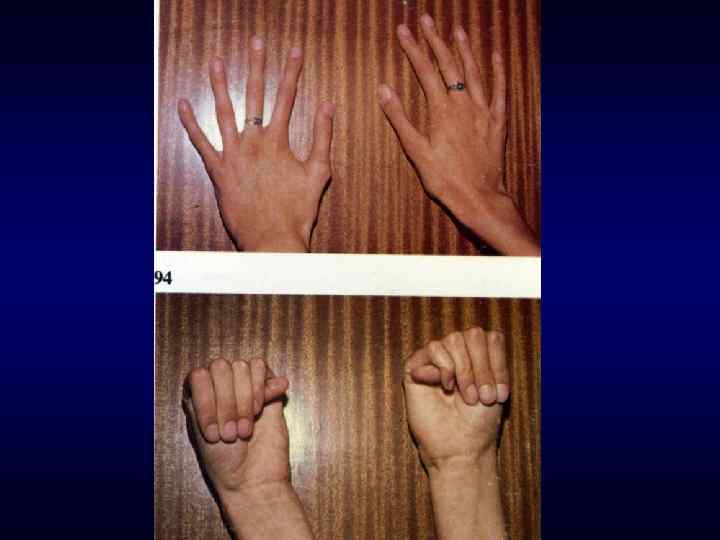
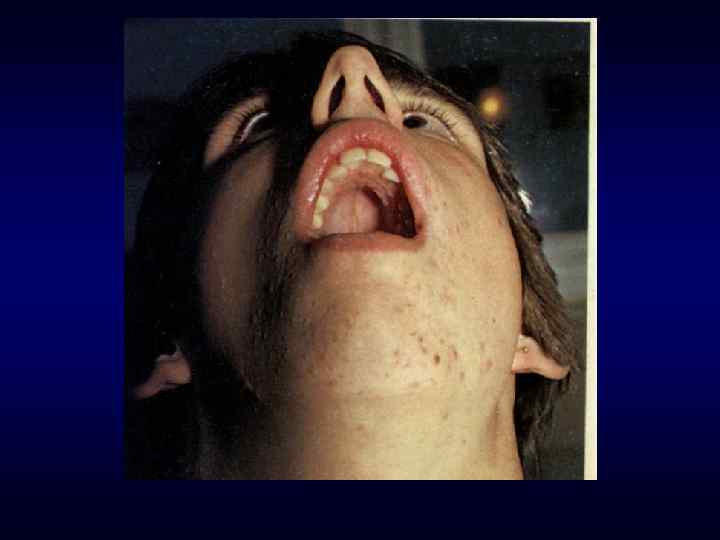
 ► Врожденный характер заболеваний ► Резистентность к") Семиотика наследственных заболеваний (общие особенности клинических проявлений) ► Врожденный характер заболеваний ► Резистентность к наиболее распространенным методам терапии. ► Фенотипических особенности индивида ►Антропометрия. ►Наличие врожденных пороков развития. ►Наличие микро аномалий развития / врожденных морфогенетических вариант.
Семиотика наследственных заболеваний (общие особенности клинических проявлений) ► Врожденный характер заболеваний ► Резистентность к наиболее распространенным методам терапии. ► Фенотипических особенности индивида ►Антропометрия. ►Наличие врожденных пороков развития. ►Наличие микро аномалий развития / врожденных морфогенетических вариант.
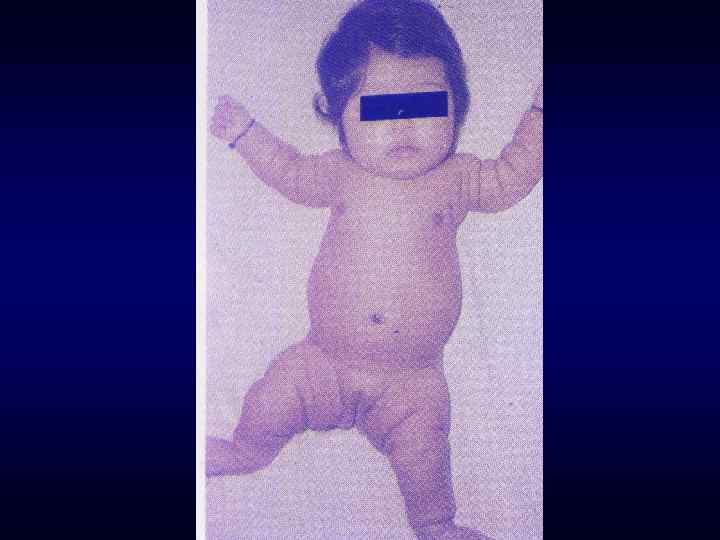
 ► Резистентность к наиболее распространенным методам терапии.") Семиотика наследственных заболеваний (общие особенности клинических проявлений) ► Резистентность к наиболее распространенным методам терапии. ► Фенотипических особенности индивида ►Антропометрия. ►Наличие врожденных пороков развития. ►Наличие микро аномалий развития / врожденных морфогенетических вариант.
Семиотика наследственных заболеваний (общие особенности клинических проявлений) ► Резистентность к наиболее распространенным методам терапии. ► Фенотипических особенности индивида ►Антропометрия. ►Наличие врожденных пороков развития. ►Наличие микро аномалий развития / врожденных морфогенетических вариант.

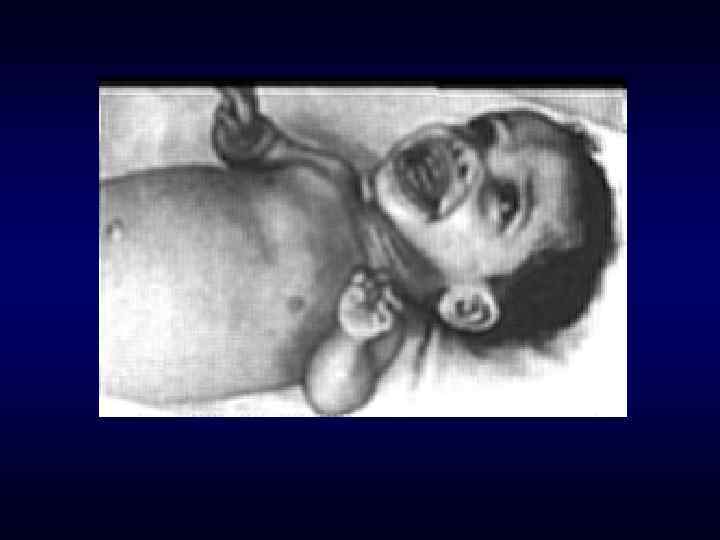
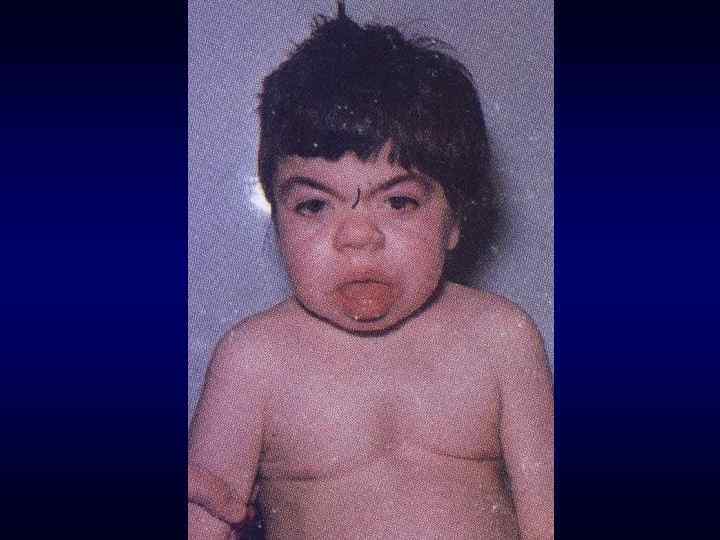
 ► Фенотипических особенности индивида ►Антропометрия. ►Наличие врожденных") Семиотика наследственных заболеваний (общие особенности клинических проявлений) ► Фенотипических особенности индивида ►Антропометрия. ►Наличие врожденных пороков развития. ►Наличие микро аномалий развития / врожденных морфогенетических вариант.
Семиотика наследственных заболеваний (общие особенности клинических проявлений) ► Фенотипических особенности индивида ►Антропометрия. ►Наличие врожденных пороков развития. ►Наличие микро аномалий развития / врожденных морфогенетических вариант.
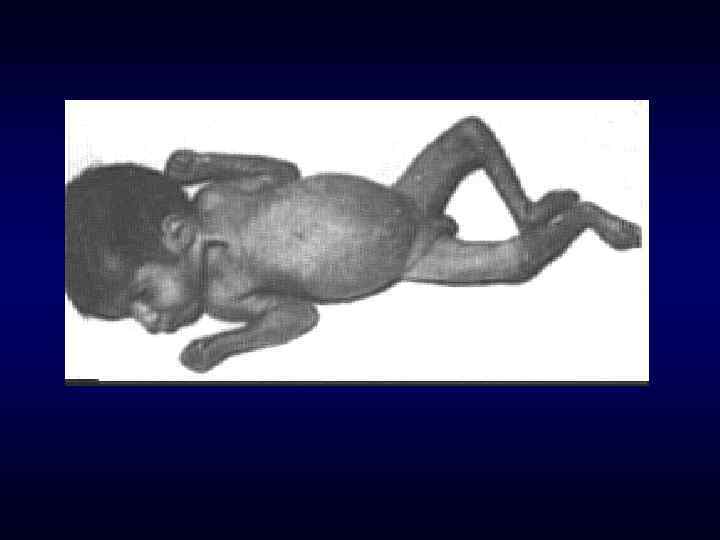
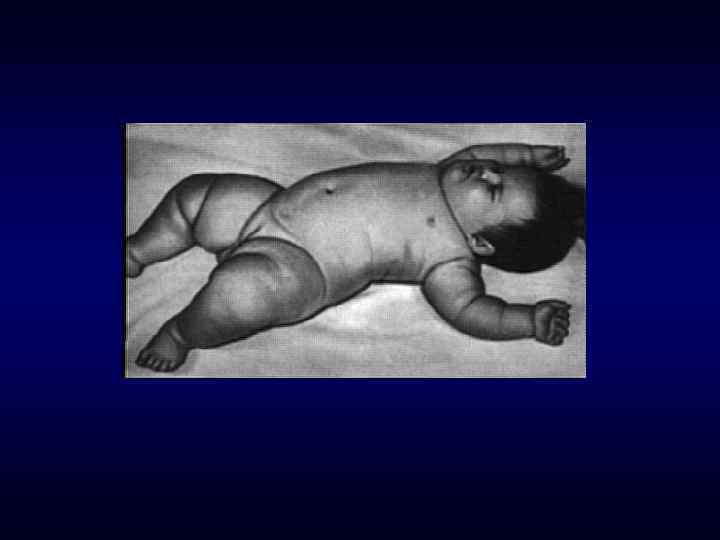
 ► Фенотипических особенности индивида ►Наличие врожденных пороков") Семиотика наследственных заболеваний (общие особенности клинических проявлений) ► Фенотипических особенности индивида ►Наличие врожденных пороков развития. ►Наличие микро аномалий развития / врожденных морфогенетических вариант.
Семиотика наследственных заболеваний (общие особенности клинических проявлений) ► Фенотипических особенности индивида ►Наличие врожденных пороков развития. ►Наличие микро аномалий развития / врожденных морфогенетических вариант.
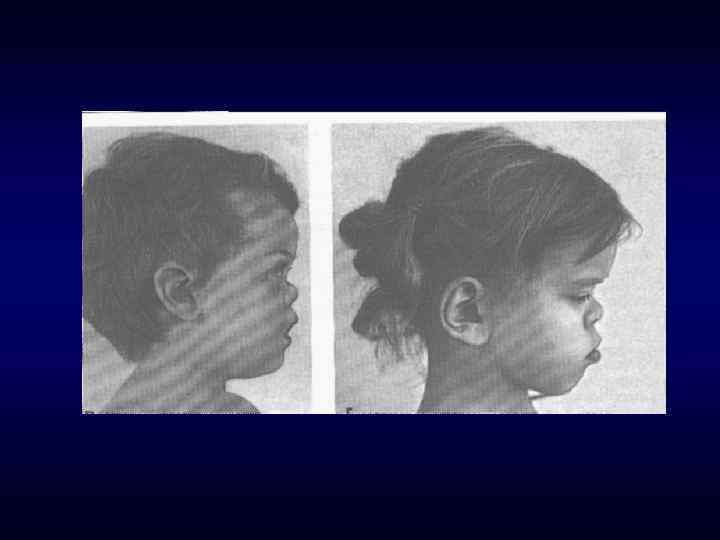
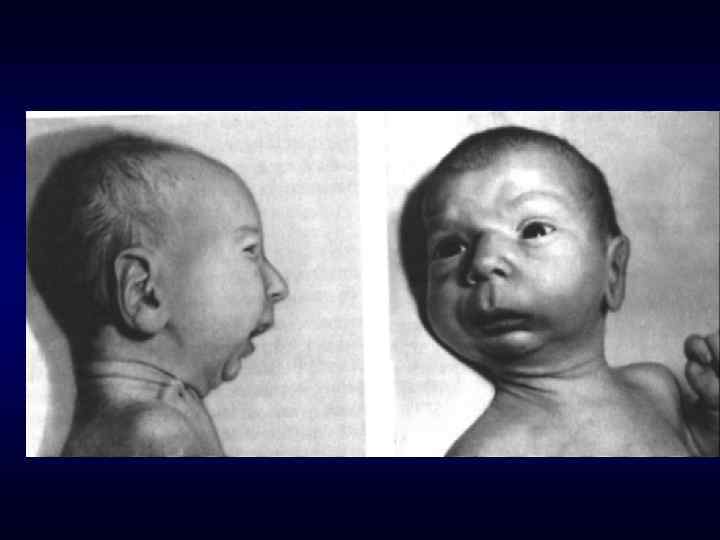
 долихоцефалия
долихоцефалия
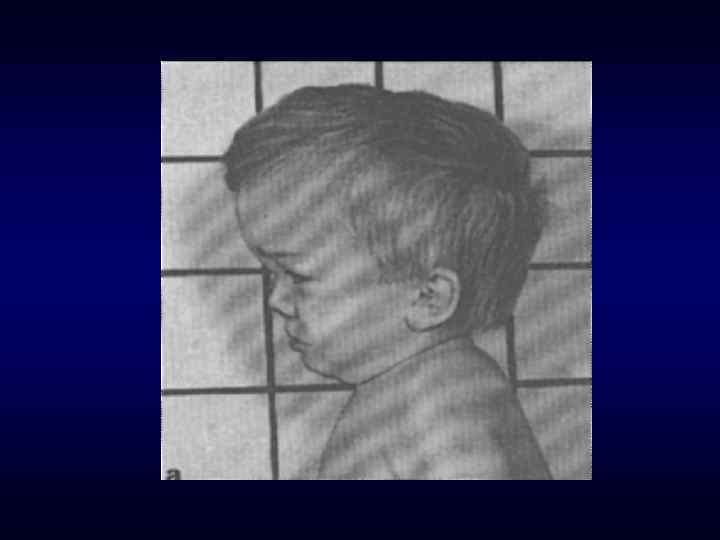
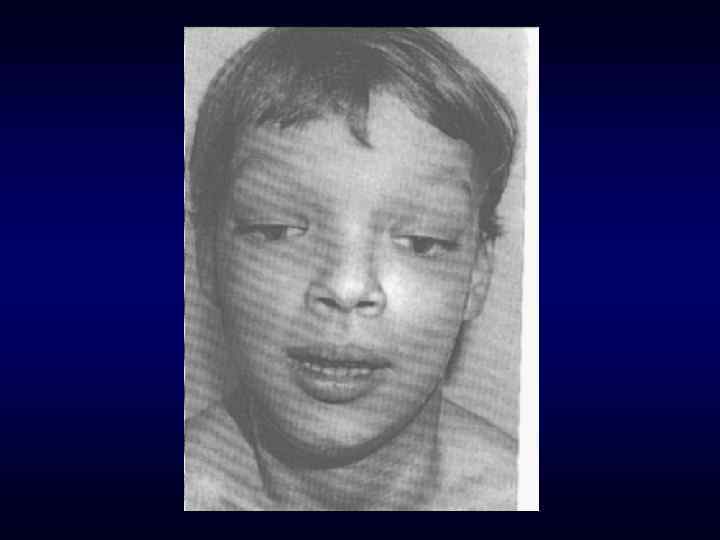
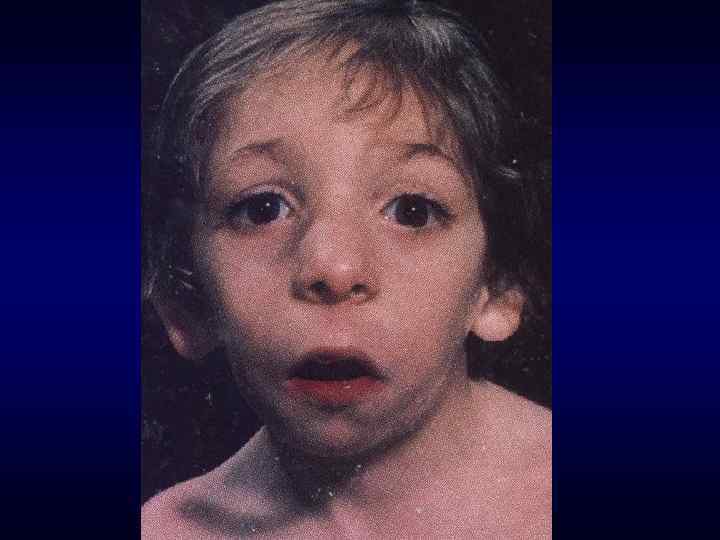
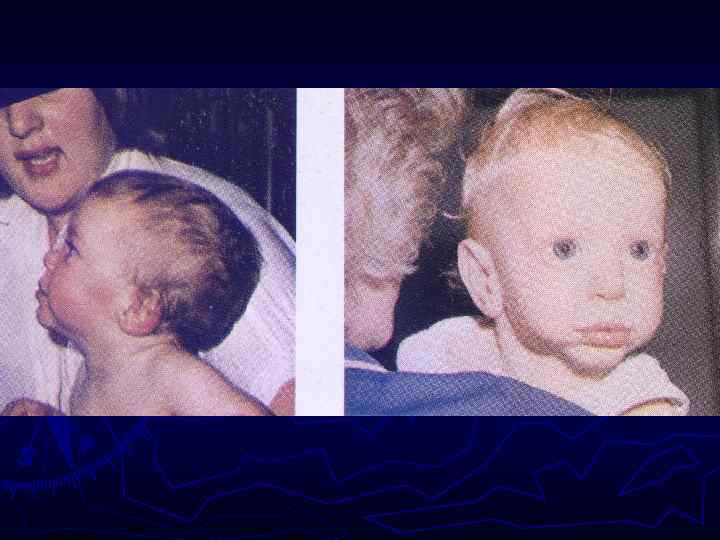
 Череп в форме трилистника
Череп в форме трилистника
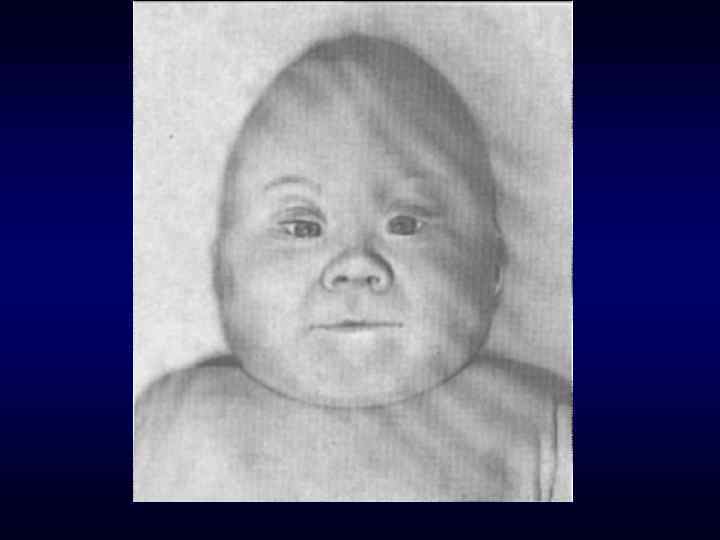
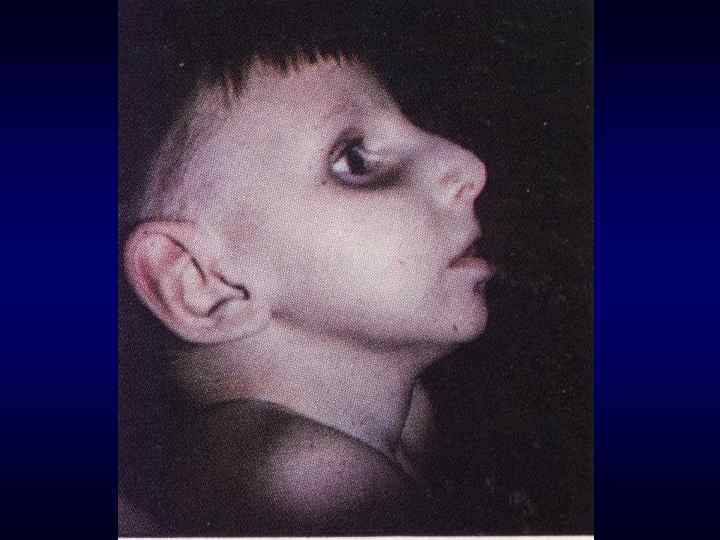
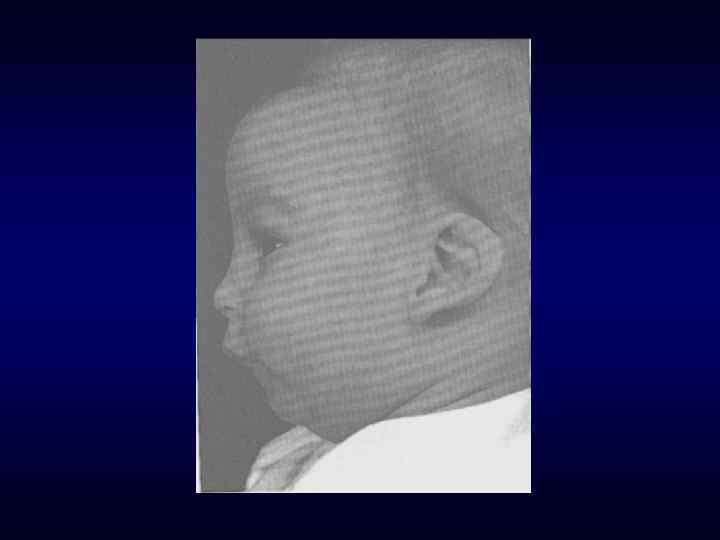
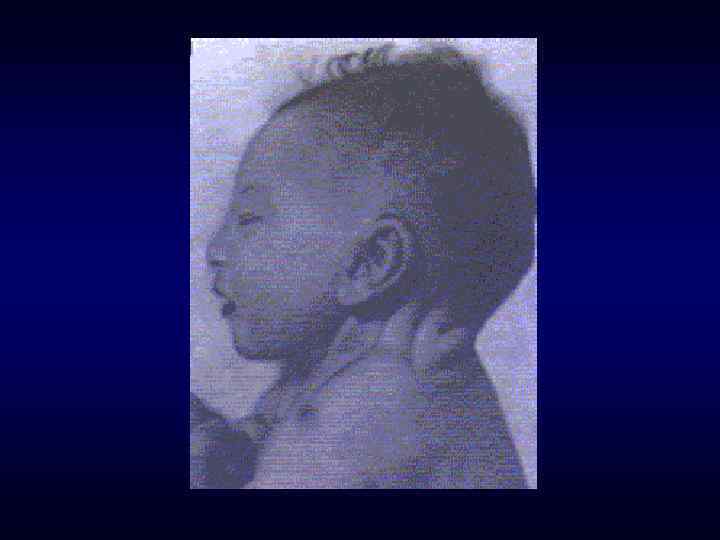
 микроцефалия
микроцефалия
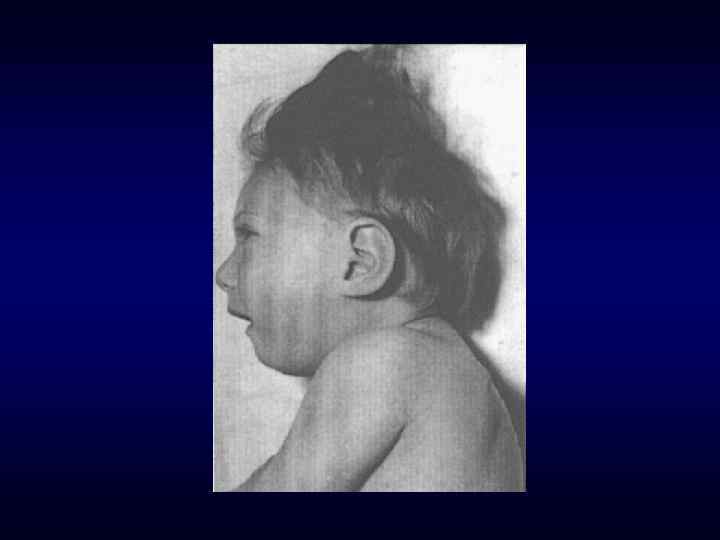
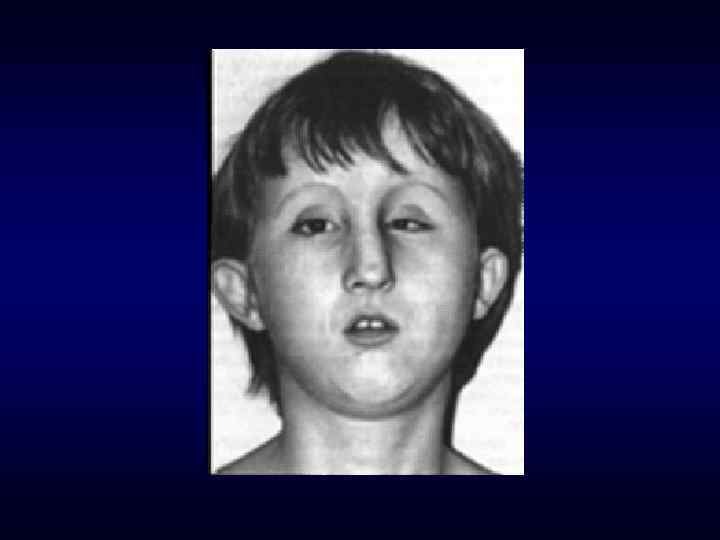
 брахицефалия
брахицефалия
 брахицефалия
брахицефалия
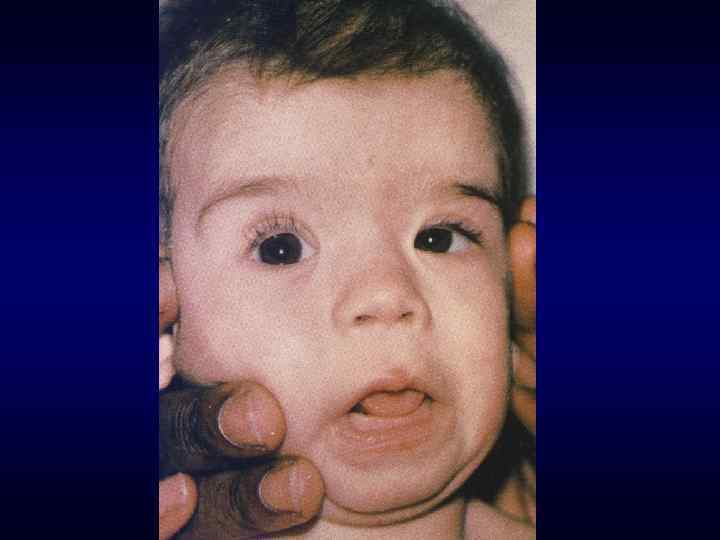
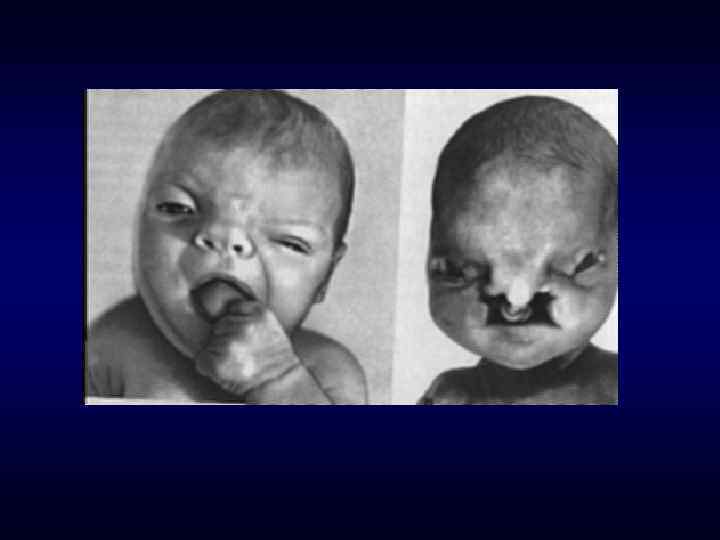
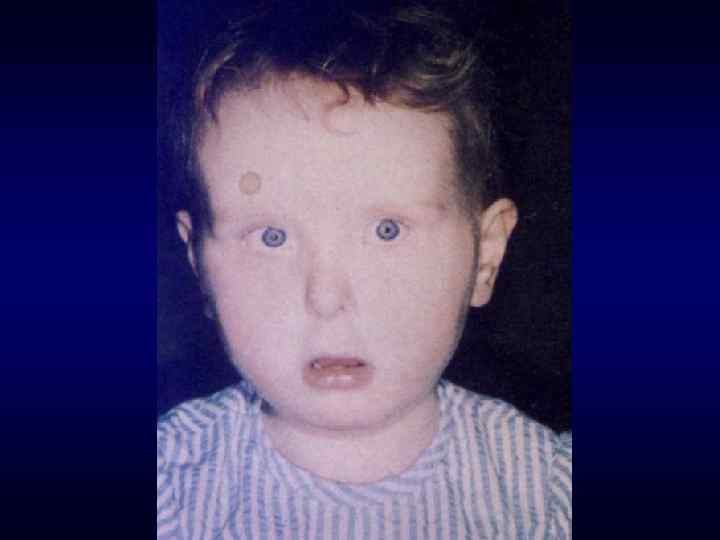
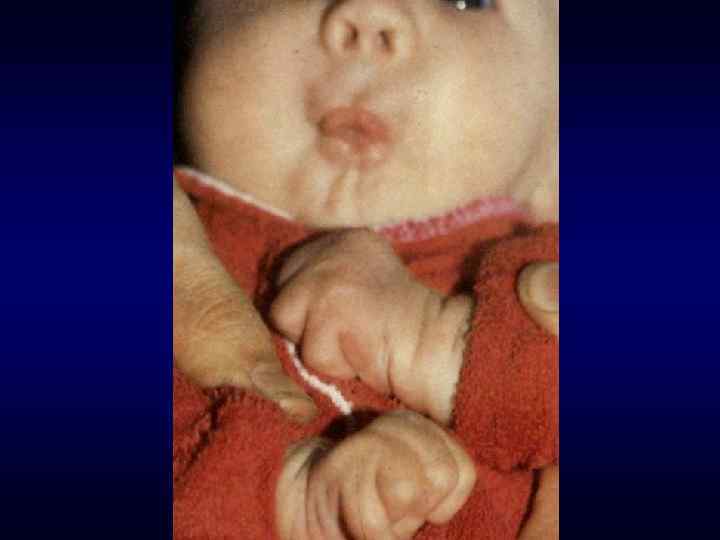
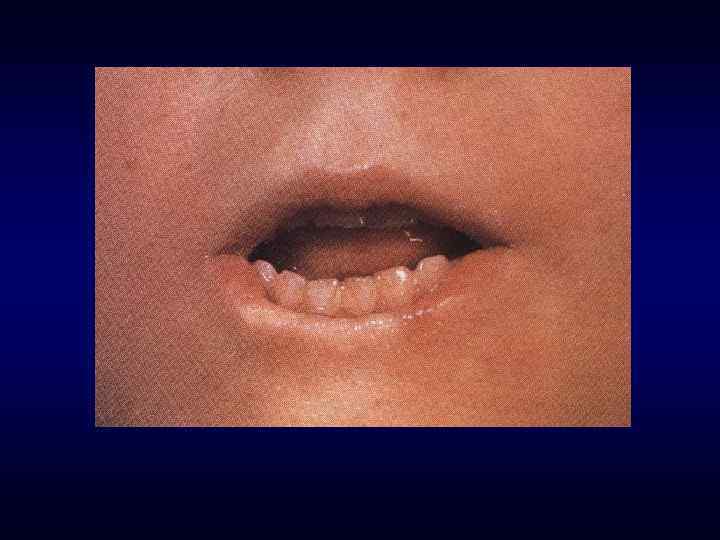
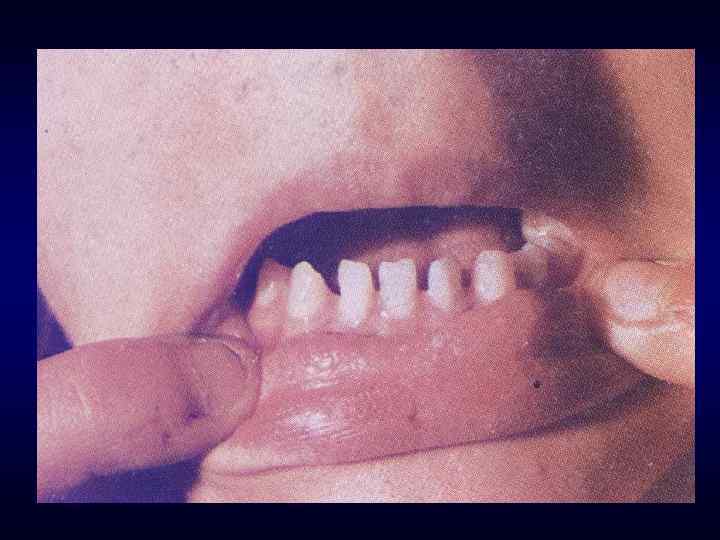
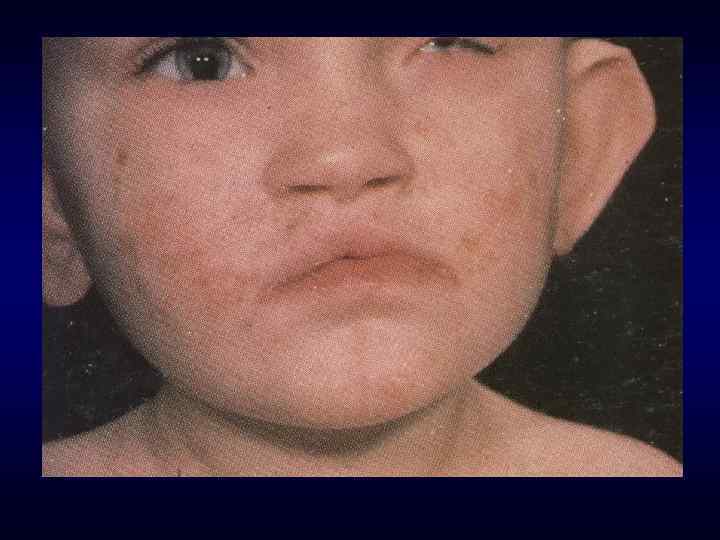
 Микрофтальм, эпикант
Микрофтальм, эпикант



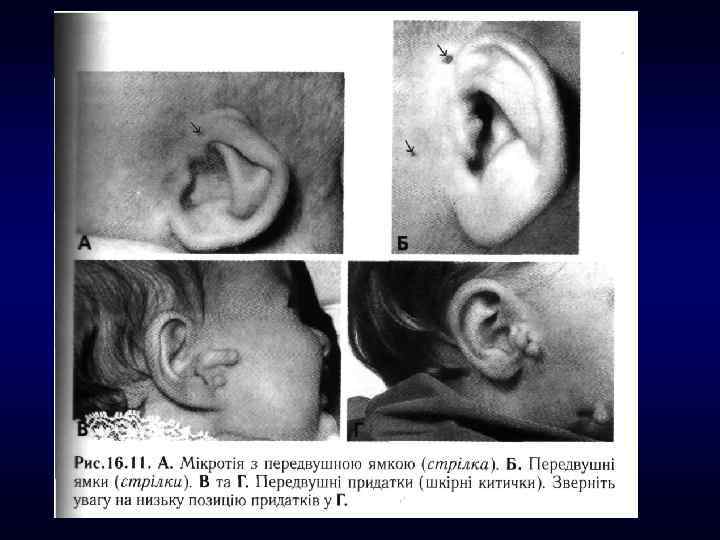
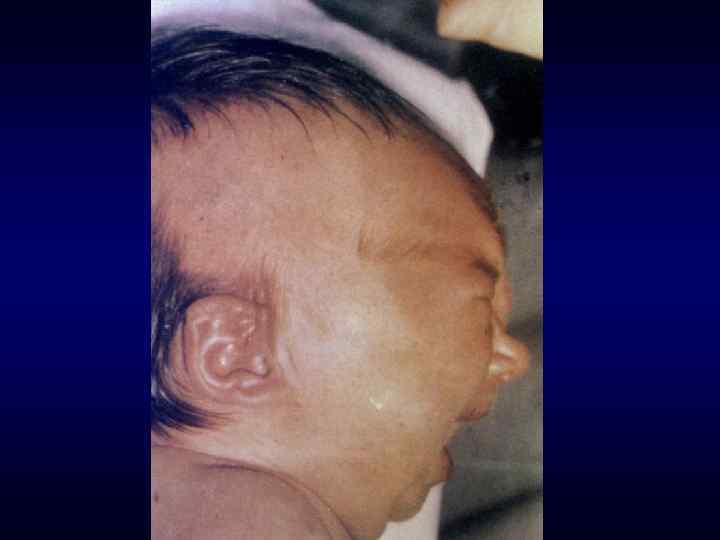
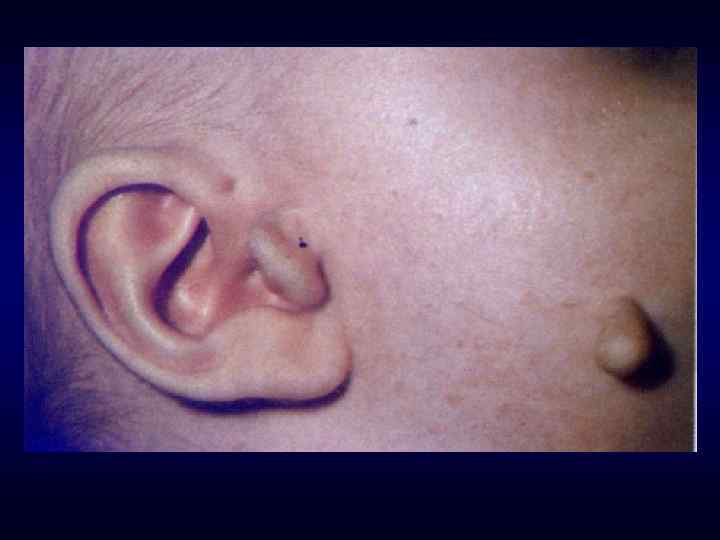
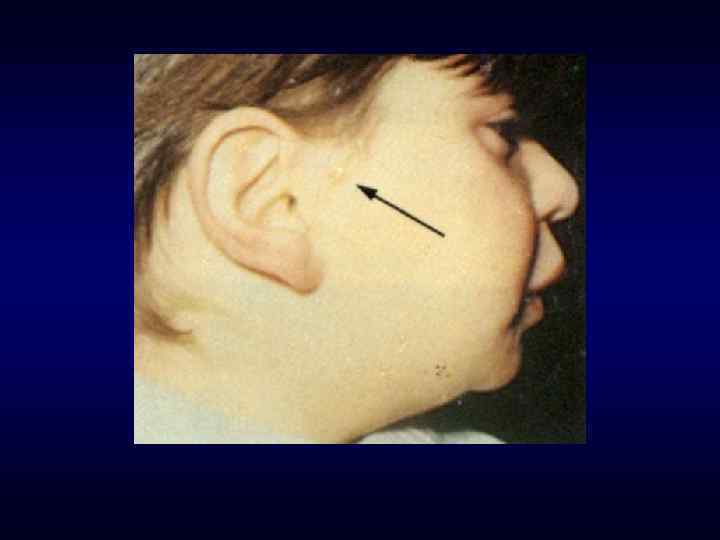

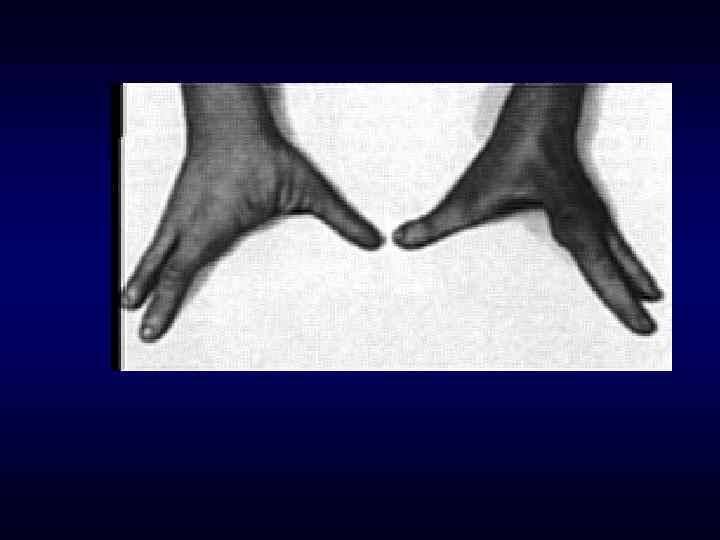
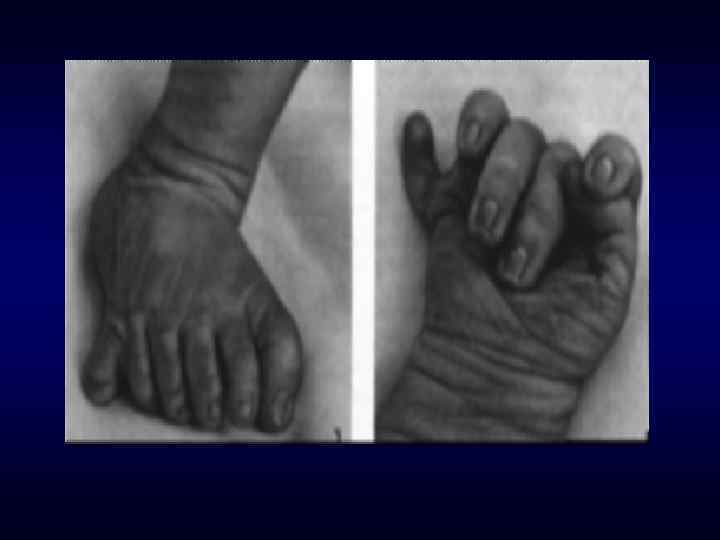
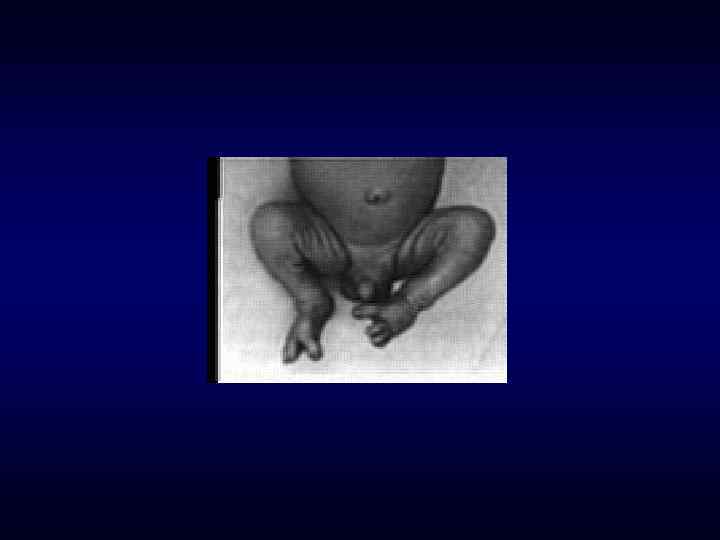
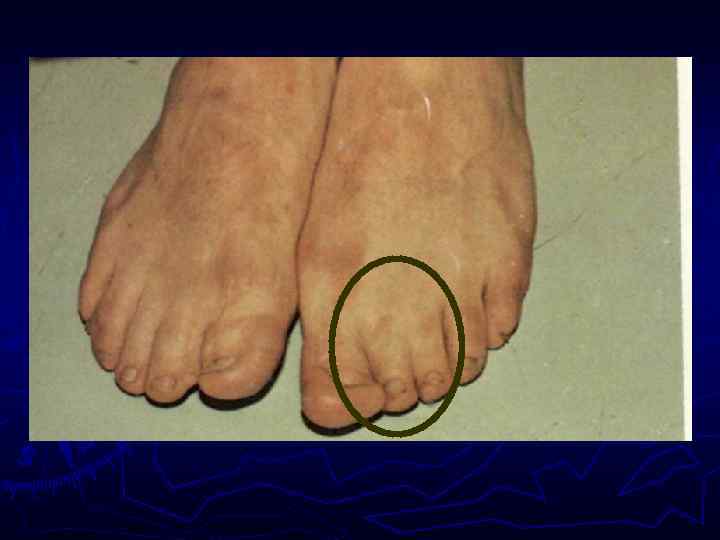
 ►- специализированная помощь населению по предупреждению появления в семье больных с") Медико-генетическое консультирование (МГК) ►- специализированная помощь населению по предупреждению появления в семье больных с наследственной патологией. ► - коммуникативный процесс, в результате которого активно заинтересованные пациенты с наследственными заболеваниями получают сведения о характере данного заболевания, а также способах его предупреждения и лечения.
Медико-генетическое консультирование (МГК) ►- специализированная помощь населению по предупреждению появления в семье больных с наследственной патологией. ► - коммуникативный процесс, в результате которого активно заинтересованные пациенты с наследственными заболеваниями получают сведения о характере данного заболевания, а также способах его предупреждения и лечения.
 Основные задачи МГК: - Установление точного диагноза наследственного заболевания. ► - Определение типа наследования заболевания в данной семье. ► - Составление прогноза рождения ребенка с наследственной болезнью. ► - Расчет риска повторения болезни в семье. ► - Определение наиболее эффективного способа профилактики. ► - Помощь семье в принятии правильного решения. ► - Пропаганда медико-генетических знаний среди врачей, населения. ►
Основные задачи МГК: - Установление точного диагноза наследственного заболевания. ► - Определение типа наследования заболевания в данной семье. ► - Составление прогноза рождения ребенка с наследственной болезнью. ► - Расчет риска повторения болезни в семье. ► - Определение наиболее эффективного способа профилактики. ► - Помощь семье в принятии правильного решения. ► - Пропаганда медико-генетических знаний среди врачей, населения. ►
 установленная или подозреваемая наследственная болезнь в семье рождение") Показаниями к МГК есть: ► 1) установленная или подозреваемая наследственная болезнь в семье рождение ребенка с врожденным пороком развития; задержка физического развития или умственная отсталость у ребенка; аномалии полового развития; ► 2) повторные спонтанные аборты, выкидыши, мертворождения; выявление патологии в ходе просеивающих программ; ► 3) кровнородственные браки; ► 4) воздействие известных или возможных тератогенов в первые 3 месяца беременности; ► 5) неблагополучное протекание беременности.
Показаниями к МГК есть: ► 1) установленная или подозреваемая наследственная болезнь в семье рождение ребенка с врожденным пороком развития; задержка физического развития или умственная отсталость у ребенка; аномалии полового развития; ► 2) повторные спонтанные аборты, выкидыши, мертворождения; выявление патологии в ходе просеивающих программ; ► 3) кровнородственные браки; ► 4) воздействие известных или возможных тератогенов в первые 3 месяца беременности; ► 5) неблагополучное протекание беременности.
 Факторы повышенного риска рождения детей с хромосомными болезнями: 1. Потомство с трисомией появляется у одних и тех же женщин повторно с частотой не менее 1%. ► 2. Родственники пробанда с трисомией 21 или другими анеуплоидиями имеют несколько повышенный риск рождения ребенка с анеуплоидией. ► 3. Кровное родство родителей может повысить риск трисомии у потомства. ► 4. Резко повышается риск рождения ребенка с трисомией у матери, чей возраст превышает 35 лет. После 45 лет каждая 5 беременность завершается рождением ребенка с хромосомной болезнью. ►
Факторы повышенного риска рождения детей с хромосомными болезнями: 1. Потомство с трисомией появляется у одних и тех же женщин повторно с частотой не менее 1%. ► 2. Родственники пробанда с трисомией 21 или другими анеуплоидиями имеют несколько повышенный риск рождения ребенка с анеуплоидией. ► 3. Кровное родство родителей может повысить риск трисомии у потомства. ► 4. Резко повышается риск рождения ребенка с трисомией у матери, чей возраст превышает 35 лет. После 45 лет каждая 5 беременность завершается рождением ребенка с хромосомной болезнью. ►
 ► Уточнение диагноза проводится с помощью метода составления родословных – генеалогического анализа. Суть метода в выявлении родственных связей и прослеживание признака или заболевания среди родственников. ► Генеалогический метод — метод родословных, т. е. прослеживание болезни (или признака) в семье или роду с указанием типа родственных связей между членами родословной.
► Уточнение диагноза проводится с помощью метода составления родословных – генеалогического анализа. Суть метода в выявлении родственных связей и прослеживание признака или заболевания среди родственников. ► Генеалогический метод — метод родословных, т. е. прослеживание болезни (или признака) в семье или роду с указанием типа родственных связей между членами родословной.
для установления наследственного характера") Генеалогический метод относится к наиболее универсальным методам в медицинской генетике. 1)для установления наследственного характера признака; 2) при определении типа наследования и пенетрантности гена; 3) при анализе сцепления генов и картировании хромосом; 4) при изучении интенсивности мутационного процесса; 5) при расшифровке механизмов взаимодействия генов; 6) при медико-генетическом консультировании.
Генеалогический метод относится к наиболее универсальным методам в медицинской генетике. 1)для установления наследственного характера признака; 2) при определении типа наследования и пенетрантности гена; 3) при анализе сцепления генов и картировании хромосом; 4) при изучении интенсивности мутационного процесса; 5) при расшифровке механизмов взаимодействия генов; 6) при медико-генетическом консультировании.
 Методы диагностики наследственных болезней ► ЦИТОГЕНЕТИЧЕСКИЕ МЕТОДЫ ► БИОХИМИЧЕСКИЕ ИССЛЕДОВАНИЯ ► МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ
Методы диагностики наследственных болезней ► ЦИТОГЕНЕТИЧЕСКИЕ МЕТОДЫ ► БИОХИМИЧЕСКИЕ ИССЛЕДОВАНИЯ ► МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ
 Показания к пренатальной диагностике ► возраст матери старше 35 лет ► наличие у предшествующего ребенка б. Дауна, хромосомной, генной патологии, ВПР ► носительство хромосомной транслокации ► гетерозиготное состояние по гену наследственного дефекта обмена.
Показания к пренатальной диагностике ► возраст матери старше 35 лет ► наличие у предшествующего ребенка б. Дауна, хромосомной, генной патологии, ВПР ► носительство хромосомной транслокации ► гетерозиготное состояние по гену наследственного дефекта обмена.
 умственная отсталость, психические нарушения; 2)") Показания для биохимического исследования ► ► ► ► 1) умственная отсталость, психические нарушения; 2) нарушение физического развития - аномальный рост и строение волос или ногтей; неправильный рост с искривлением костей туловища и конечностей, чрезмерное отло-жение жира, гипотрофия или кахексия, тугоподвижность или разболтанность суставов; 3) плохое зрение или полная слепота, тугоухость или глухота; 4) судороги, мышечная гипотония, гипер- и гипопигментация, фото-чувствительность, желтуха; 5) непереносимость отдельных пищевых продуктов и лекарственных препаратов, нарушение пищеварения, частая рвота, диарея, жидкий стул, гепато- и спленомегалия; 6) почечно-каменная болезнь, холестаз; 7) гемолитические анемии и др. состояния.
Показания для биохимического исследования ► ► ► ► 1) умственная отсталость, психические нарушения; 2) нарушение физического развития - аномальный рост и строение волос или ногтей; неправильный рост с искривлением костей туловища и конечностей, чрезмерное отло-жение жира, гипотрофия или кахексия, тугоподвижность или разболтанность суставов; 3) плохое зрение или полная слепота, тугоухость или глухота; 4) судороги, мышечная гипотония, гипер- и гипопигментация, фото-чувствительность, желтуха; 5) непереносимость отдельных пищевых продуктов и лекарственных препаратов, нарушение пищеварения, частая рвота, диарея, жидкий стул, гепато- и спленомегалия; 6) почечно-каменная болезнь, холестаз; 7) гемолитические анемии и др. состояния.
 С помощью цитогенетического метода определяется наличие Х и У полового хроматина, определяющего истинную половую принадлежность. Показания для цитогенетического обследования больного: ► ► ► ► ► 1) множественные пороки развития (с вовлечением трех и более систем); наиболее постоянные нарушения - пороки рзвития головного мозга, опорнодвигательной системы, сердца и мочеполовой системы; 2) умственная отсталость в сочетании с нарушениями физического развития, дисплазиями, гипогенитализмом; 3) стойкое первичное бесплодие у мужчин и у женщин при исключении гинекологической и урологической патологии; 4) привычное невынашивание беременности, особенно на ранних стадиях; 5) нарушение полового развития (гипогонадизм, половые инверсии); 6) небольшая масса ребенка, рожденного при доношенной беременности. Применение цитогенетического метода в клинической генетике обусловило развитие нового направления - клинической цитогенетики, которая позволяет: - установить происхождение структурно перестроенных хромосом и их точную классификацию; - выделить синдромы, обусловленные дисбалансом по участкам индивидуальных хромосом; - накапливать сведения об изменениях хромосом в опухолевых клетках, у больных с наследственными заболеваниями крови и т. д.
С помощью цитогенетического метода определяется наличие Х и У полового хроматина, определяющего истинную половую принадлежность. Показания для цитогенетического обследования больного: ► ► ► ► ► 1) множественные пороки развития (с вовлечением трех и более систем); наиболее постоянные нарушения - пороки рзвития головного мозга, опорнодвигательной системы, сердца и мочеполовой системы; 2) умственная отсталость в сочетании с нарушениями физического развития, дисплазиями, гипогенитализмом; 3) стойкое первичное бесплодие у мужчин и у женщин при исключении гинекологической и урологической патологии; 4) привычное невынашивание беременности, особенно на ранних стадиях; 5) нарушение полового развития (гипогонадизм, половые инверсии); 6) небольшая масса ребенка, рожденного при доношенной беременности. Применение цитогенетического метода в клинической генетике обусловило развитие нового направления - клинической цитогенетики, которая позволяет: - установить происхождение структурно перестроенных хромосом и их точную классификацию; - выделить синдромы, обусловленные дисбалансом по участкам индивидуальных хромосом; - накапливать сведения об изменениях хромосом в опухолевых клетках, у больных с наследственными заболеваниями крови и т. д.
; ► инвазивные (биопсия хориона -") Методы пренатальной диагностики ► неинвазивные (УЗИ, фетоскопия, определение фетопротеина); ► инвазивные (биопсия хориона - хориоцентез с культивированием клеток, амниоцентез с цитологическим, цитогенетическим, биохимическим, молекулярно - генетическим изучением клеток амниона и околоплодной жидкости, кордоцентез – пункция вены пуповины исследование крови плода, биопсия клеток плода).
Методы пренатальной диагностики ► неинвазивные (УЗИ, фетоскопия, определение фетопротеина); ► инвазивные (биопсия хориона - хориоцентез с культивированием клеток, амниоцентез с цитологическим, цитогенетическим, биохимическим, молекулярно - генетическим изучением клеток амниона и околоплодной жидкости, кордоцентез – пункция вены пуповины исследование крови плода, биопсия клеток плода).
 МОЛЕКУЛЯРНО-ЦИТОГЕНЕТИЧЕСКИЕ И МОЛЕКУЛЯРНО-БИОЛОГИЧЕСКИЕ МЕТОДЫ Это большая и разнообразная группа методов исследования молекулярной структуры ДНК, основные дифференциальнодиагностические тесты, необходимость разработки которых обусловлена генетической природой наследственных заболеваний, их выраженным клиническим полиморфизмом. Особое место в этой группе занимают методы ДНК-диагностики (зондовой). Они позволяют диагностировать заболевание на уровне первичного молекулярного дефекта - патологического гена. Ее точность в установлении причины наследственного дефекта абсолютна. ► Среди основных методов ДНК-диагностики выделяются: - дозовый блот-гибридизационный анализ; - анализ полиморфизма длин рестрикционных фрагментов (ПДРФ); - полимеразная цепная реакция (ПЦР); - анализ полиморфизма микросателлитных последовательностей. ► ► Благодаря этим методам у врачей появились уникальные возможности эффективного применения в различных областях медицины самых совершенных технологий.
МОЛЕКУЛЯРНО-ЦИТОГЕНЕТИЧЕСКИЕ И МОЛЕКУЛЯРНО-БИОЛОГИЧЕСКИЕ МЕТОДЫ Это большая и разнообразная группа методов исследования молекулярной структуры ДНК, основные дифференциальнодиагностические тесты, необходимость разработки которых обусловлена генетической природой наследственных заболеваний, их выраженным клиническим полиморфизмом. Особое место в этой группе занимают методы ДНК-диагностики (зондовой). Они позволяют диагностировать заболевание на уровне первичного молекулярного дефекта - патологического гена. Ее точность в установлении причины наследственного дефекта абсолютна. ► Среди основных методов ДНК-диагностики выделяются: - дозовый блот-гибридизационный анализ; - анализ полиморфизма длин рестрикционных фрагментов (ПДРФ); - полимеразная цепная реакция (ПЦР); - анализ полиморфизма микросателлитных последовательностей. ► ► Благодаря этим методам у врачей появились уникальные возможности эффективного применения в различных областях медицины самых совершенных технологий.
 Различают три вида профилактики наследственной патологии. Первичная профилактика ► Под первичной профилактикой понимают такие действия, которые должны предупредить зачатие больного ребёнка. ► Реализуется это планированием деторождения и улучшением среды обитания человека.
Различают три вида профилактики наследственной патологии. Первичная профилактика ► Под первичной профилактикой понимают такие действия, которые должны предупредить зачатие больного ребёнка. ► Реализуется это планированием деторождения и улучшением среды обитания человека.
 Планирование деторождения включает три основные позиции. 1. Оптимальный репродуктивный возраст, который для женщин составляет 21— 35 лет (более ранние или поздние беременности увеличивают вероятность рождения ребёнка с врождённой патологией и с хромосомными болезнями) ► 2. Отказ от деторождения в случаях высокого риска наследственной и врождённой патологии (при отсутствии надёжных методов дородовой диагностики, лечения, адаптации и реабилитации больных). ► 3. Отказ от деторождения в браках с кровными родственниками и между двумя гетерозиготными носителями патологического гена ►
Планирование деторождения включает три основные позиции. 1. Оптимальный репродуктивный возраст, который для женщин составляет 21— 35 лет (более ранние или поздние беременности увеличивают вероятность рождения ребёнка с врождённой патологией и с хромосомными болезнями) ► 2. Отказ от деторождения в случаях высокого риска наследственной и врождённой патологии (при отсутствии надёжных методов дородовой диагностики, лечения, адаптации и реабилитации больных). ► 3. Отказ от деторождения в браках с кровными родственниками и между двумя гетерозиготными носителями патологического гена ►
 ► Вторичная профилактика осуществляется путём прерывания беременности в случае высокой вероятности заболевания плода или пренатально диагностированной болезни. ► Прерывание можно делать только в установленные сроки и с согласия женщины. Основанием для элиминации эмбриона или плода является наследственная болезнь.
► Вторичная профилактика осуществляется путём прерывания беременности в случае высокой вероятности заболевания плода или пренатально диагностированной болезни. ► Прерывание можно делать только в установленные сроки и с согласия женщины. Основанием для элиминации эмбриона или плода является наследственная болезнь.
 ► Под третичной профилактикой наследственной патологии понимают коррекцию проявления патологических генотипов. Это можно назвать и нормокопированием. поскольку при патологическом генотипе стремятся получить нормальный фенотип. ► Третичная профилактика применяется как при наследственных болезнях, так и (особенно часто) при болезнях с наследственной предрасположенностью. С её помощью можно добиться полной нормализации или снижения выраженности патологического процесса.
► Под третичной профилактикой наследственной патологии понимают коррекцию проявления патологических генотипов. Это можно назвать и нормокопированием. поскольку при патологическом генотипе стремятся получить нормальный фенотип. ► Третичная профилактика применяется как при наследственных болезнях, так и (особенно часто) при болезнях с наследственной предрасположенностью. С её помощью можно добиться полной нормализации или снижения выраженности патологического процесса.
 ПРЕКОНЦЕПЦИОННАЯ ПРОФИЛАКТИКА. ► комплекс мероприятий, потенциально способных обеспечить оптимальные условия для созревания яйцеклетки, ее последующего развития, имплантации, и как результат - развития плода. ► осуществляется в отношении врожденных пороков развития и других мультифакториальных состояний, т. е. не детерминируемых менделирующим наследованием после медико-генетического исследования семьи, в процессе которого врачгенетик определяет характер наследования заболевания, повторный генетический риск, возможную эффективность профилактики.
ПРЕКОНЦЕПЦИОННАЯ ПРОФИЛАКТИКА. ► комплекс мероприятий, потенциально способных обеспечить оптимальные условия для созревания яйцеклетки, ее последующего развития, имплантации, и как результат - развития плода. ► осуществляется в отношении врожденных пороков развития и других мультифакториальных состояний, т. е. не детерминируемых менделирующим наследованием после медико-генетического исследования семьи, в процессе которого врачгенетик определяет характер наследования заболевания, повторный генетический риск, возможную эффективность профилактики.
 у будущих") Комплекс преконцепционной профилактики включает: 1. Лечение хронических очагов инфекций (если таковые имеютсяя) у будущих родителей. 2. Лечение хронических соматических заболеваний. 3. Оценка спермограммы. 4. Регулирование полового режима, планирование беременности. 5. Диета, обогащенная витаминами и микроэлементами, в том числе фолиевой кислотой (считается, что она способствует уменьшению риска рождения ребенка с пороками ЦНС). Обычно профилактика назначается и осуществляется врачом-гинекологом, с участием других специалистов.
Комплекс преконцепционной профилактики включает: 1. Лечение хронических очагов инфекций (если таковые имеютсяя) у будущих родителей. 2. Лечение хронических соматических заболеваний. 3. Оценка спермограммы. 4. Регулирование полового режима, планирование беременности. 5. Диета, обогащенная витаминами и микроэлементами, в том числе фолиевой кислотой (считается, что она способствует уменьшению риска рождения ребенка с пороками ЦНС). Обычно профилактика назначается и осуществляется врачом-гинекологом, с участием других специалистов.
: ► ► ► ► ► - генетический") Показания к проведению преконцепционной профилактики (по Холингсворт): ► ► ► ► ► - генетический риск мультифакториальных пороков развития в семье; - повторные спонтанные аботры и рождение мертвых плодов; - рождение детей с пренатальной гипотрофией и преждевременные роды в анамнезе; - сахарный диабет, другие эндокринные и метаболические заболевания у матери; - хронические соматические заболевания у одного или обоих родителей; - профессиональные вредности у одного из супругов; - расстройства питания; - долговременное употребление лекарственных препаратов; - заболевания, вызванные TORCH-инфекциями. С генетических позиций преконцепционная профилактика это попытка устранить условия и факторы, способствующие экспрессии патологических генов.
Показания к проведению преконцепционной профилактики (по Холингсворт): ► ► ► ► ► - генетический риск мультифакториальных пороков развития в семье; - повторные спонтанные аботры и рождение мертвых плодов; - рождение детей с пренатальной гипотрофией и преждевременные роды в анамнезе; - сахарный диабет, другие эндокринные и метаболические заболевания у матери; - хронические соматические заболевания у одного или обоих родителей; - профессиональные вредности у одного из супругов; - расстройства питания; - долговременное употребление лекарственных препаратов; - заболевания, вызванные TORCH-инфекциями. С генетических позиций преконцепционная профилактика это попытка устранить условия и факторы, способствующие экспрессии патологических генов.
 программ является выявление того или иного заболевания в доклинической стадии.") Основной целью скринирующих (просеивающих) программ является выявление того или иного заболевания в доклинической стадии. ► ► ► Прежде всего это касается наследственных болезней обмена. Они включаются в программы массового просеивания, отбираются по ряду критериев (К. Д. Краснопольская, 1986): 1. Заболевания, приводящие к выраженному снижению жизне- и трудоспособности без своевременного выявления и лечения. 2. Заболевания, достаточно распространенные в популяции (частота не менее 1: 50 000 - 200 000 новорожденных). 3. Заболевания, которые поддаются лечению с достижением принципиального эффекта для пациента, для которых разработаны эффективные методы профилактики. 4. заболевания, для которых разработан адекватный просеивающий тест.
Основной целью скринирующих (просеивающих) программ является выявление того или иного заболевания в доклинической стадии. ► ► ► Прежде всего это касается наследственных болезней обмена. Они включаются в программы массового просеивания, отбираются по ряду критериев (К. Д. Краснопольская, 1986): 1. Заболевания, приводящие к выраженному снижению жизне- и трудоспособности без своевременного выявления и лечения. 2. Заболевания, достаточно распространенные в популяции (частота не менее 1: 50 000 - 200 000 новорожденных). 3. Заболевания, которые поддаются лечению с достижением принципиального эффекта для пациента, для которых разработаны эффективные методы профилактики. 4. заболевания, для которых разработан адекватный просеивающий тест.
, ►") Сегодня в числе скринируемых заболеваний: муковисцидоз (частота - 1: 1, 5 -2 000), ► врожденный гипотиреоз (1: 4, 7 -5 000), недостаточность альфа-1 -антитрипсина (1: 5 000), фенилкетонурия (1: 10 000), ► гистидинемия (1: 23 000), ► галактоземия (1: 35 -50 000), ► лейциноз (1: 90 -120 000), ► аргинин-янтарнаая ацидурия (1: 300 000), тирозинемия (1: 900 000), ► недостаточность аденозиндезаминазы (1: 1 500 000), болезнь Тея-Сакса (частота в популяции евреевашкенази - 1: 3 700) и т. д. Следует подчеркнуть, что процедура скрининга не обеспечивает окончательного диагноза, а выявляет предположительных «больных» , которым на втором этапе требуется специализированное углубленное обследование с использованием биохимических, молекулярно-генетических и клинических методов диагностики.
Сегодня в числе скринируемых заболеваний: муковисцидоз (частота - 1: 1, 5 -2 000), ► врожденный гипотиреоз (1: 4, 7 -5 000), недостаточность альфа-1 -антитрипсина (1: 5 000), фенилкетонурия (1: 10 000), ► гистидинемия (1: 23 000), ► галактоземия (1: 35 -50 000), ► лейциноз (1: 90 -120 000), ► аргинин-янтарнаая ацидурия (1: 300 000), тирозинемия (1: 900 000), ► недостаточность аденозиндезаминазы (1: 1 500 000), болезнь Тея-Сакса (частота в популяции евреевашкенази - 1: 3 700) и т. д. Следует подчеркнуть, что процедура скрининга не обеспечивает окончательного диагноза, а выявляет предположительных «больных» , которым на втором этапе требуется специализированное углубленное обследование с использованием биохимических, молекулярно-генетических и клинических методов диагностики.
 Лечение моногенных наследственных болезней ► Консервативные методы. ► Заместительная терапия недостающими гормонами, витаминами, белками. ► Медикаментозная коррекция обмена веществ. ► Генная терапия.
Лечение моногенных наследственных болезней ► Консервативные методы. ► Заместительная терапия недостающими гормонами, витаминами, белками. ► Медикаментозная коррекция обмена веществ. ► Генная терапия.
 СПАСИБО ЗА ВНИМАНИЕ!
СПАСИБО ЗА ВНИМАНИЕ!