8 Аутоиммунные болезни Амилоидоз 2012 ред.ppt
- Количество слайдов: 85


. Клиникоморфологическая")
Лекция 8 Аутоиммунизация и аутоиммунные болезни. Патогенез. Синдромы иммунного дефицита (первичные и вторичные). Клиникоморфологическая характеристика. Амилоидоз.
 называют состояние, характеризующееся появлением реакции иммунной системы на нормальные антигены собственных")
Аутоиммунизацией (аутоаллергией, аутоагрессией) называют состояние, характеризующееся появлением реакции иммунной системы на нормальные антигены собственных тканей Происходит «срыв толерантности»

Иммунологическая толерантность – это состояние, при котором иммунный ответ на специфический антиген не развивается

Механизмы иммунологической толерантности Клональная делеция – процесс элиминации клонов аутореактивных клеток Клональная анэргия – состояние функциональной ареактивности отдельных клонов лимфоцитов на специфические антигены Периферическая супрессия – ряду аутореактивных Т- и В-лимфоцитов с низкоаффинными рецепторами в процессе своего развития удается избежать отрицательной селекции. Функциональная активность таких категорий клеток сдерживается механизмами активной клеточной супрессии

Уровни иммунологической толерантности Вид толерантности Механизм Локализация Центральная Делеция Обработка Тимус Костный мозг Разделение антигенов Барьеры, препятствующие контакту Эндокринные железы (щитовидная железа) Периферическая анергия Инактивация путем слабого сигнала Периферическая лимфоидная ткань Регуляторные клетки Подавление путем контактов и цитокинов Периферическая лимфоидная ткань Цитокиновое влияние Дифференцировка в направлении Th 2 Периферическая лимфоидная ткань Клональная делеция Постактивационный апоптоз Периферическая лимфоидная ткань

Признаки аутоиммунных заболеваний: • Наличие иммунной реакции • Наличие данных о первичности аутоиммунной реакции • Отсутствие других определенных причин болезни

Признаки аутоиммунных заболеваний: • Семейный анамнез в отношении данного заболевания или заболевания, относящегося к аутоиммунным • Наличие у данного больного другого аутоиммунного заболевания • Обнаружение мононуклеарной клеточной инфильтрации в пораженных органах и тканях • Наличие определенных генов главного комплекса гистосовместимости 2 класса (MHC class II) • Высокий уровень аутоантител, относящихся к классу Ig. G • Отложение иммунных комплексов в пораженных органах и тканях • Улучшение клинических проявлений после назначения иммуносупрессивной терапии (например, кортикостероидов)
 модификация молекул (гаптен), б) молекулярная мимикрия")
Механизмы аутоиммунных болезней Обходной путь толерантности Т-хелперов а) модификация молекул (гаптен), б) молекулярная мимикрия (перекрестный иммунитет) Поликлональная активация лимфоцитов при преобладании клональной анергии при иммунотолерантности Дисбаланс Т-супрессоров и Т-хелперов СКВ – подавление Т-супрессоров и активация Т-хелперов Появление секвестрированного антигена попадание изолированного АГ в кровоток при травме (в т. ч. психической) Генетические факторы иммунитета семейная предрасположенность, связь с HLA-антигенами Микробные агенты в аутоиммунитете митогенный, супрессорный эффект, перекрестный иммунитет
")
Повреждение барьеров Т-клетка Барьер Забарьерная клетка-мишень (распознается как чужеродная)

Экстраординарная экспрессия костимулирующих молекул В 7 Клетка-стимулятор и мишень (наличие на ее поверхности костимулятора В 7 делает ее аутоантигенной для Т-клеток)

Антигенная мимикрия Антитело Микроорганизм Аутологичная клетка-мишень, несущая антигены, сходные с антигеном микроорганизма

Модификация аутоантителами Модифицирующий агент Аутологичная клетка Модифицированные аутологичные клетки - мишени аутоантител

Эффекты, опосредованные аутоантителами С'-зависимый лизис

Эффекты, опосредованные Тклетками Воспаление, деструкция

Этиология и патогенез аутоиммунных болезней Предрасполагающие Инициирующие генетические факторы: факторы - биологические - химические - физические Способствующие факторы: - дисфункция иммунной системы Аутоиммунные болезни

Аутоиммунные болезни Группы болезней Болезни 1. Аутоиммунные болезни нервной системы а. Энцефаломиелит; б. Полиневрит в. Рассеянный склероз; г. Симпатическая офтальмия Органоспецифические аутоиммунные болезни 2. Аутоиммунные заболевания желез внутренней секреции а. Аутоиммунный гипотиреоидит б. Тиреоидиты (гиперпластический и атрофический) в. Тиреотоксикоз; г. Первичная микседема д. Гипер-и гипопаратиреоз; е. Сахарный диабет I типа ж Аддисонова болезнь (аутоиммунизация); з. Асперматогения 3. Аутоиммунные болезни крови а. Гипопластическая анемия; Органонеспецифические аутоиммунные болезни Аутоиммунные болезни промежуточного типа б. Апластическая анемия 1. Системная красная волчанка 2. Ревматоидный артрит 3. Системная склеродермия 4. Дерматомиозит 5. Тромбоцитопеническая пурпура 1. Синдром Шегрена 2. Синдром Гудпасчера 3. Аутоиммунный гастрит типа А 4. Миастения Гравис 5. Первичный билиарный цирроз печени

Аутоиммунные болезни I группа. Органоспецифические аутоиммунные болезни 1. Аутоиммунные болезни нервной системы а. Энцефаломиелит б. Полиневрит в. Рассеянный склероз г. Симпатическая офтальмия

Аутоиммунные болезни I группа. Органоспецифические аутоиммунные болезни 2. Аутоиммунные заболевания желез внутренней секреции а. Аутоиммунный гипотиреоидит б. Тиреоидиты (гиперпластический и атрофический) в. Тиреотоксикоз г. Первичная микседема д. Гипер-и гипопаратиреоз е. Сахарный диабет I типа ж. Аддисонова болезнь (аутоиммунизация) з. Асперматогения

Аутоиммунные болезни I группа. Органоспецифические аутоиммунные болезни 3. Аутоиммунные болезни крови а. Гипопластическая анемия б. Апластическая анемия

Аутоиммунные болезни II группа. Органонеспецифические аутоиммунные болезни а. Системная красная волчанка б. Ревматоидный артрит в. Системная склеродермия г. Дерматомиозит д. Тромбоцитопеническая пурпура

Аутоиммунные болезни III группа. Аутоиммунные болезни промежуточного типа а. Синдром Шегрена б. Синдром Гудпасчера в. Аутоиммунный гастрит типа А г. Миастения Гравис д. Первичный билиарный цирроз печени
 • Распространенность – до 1 на 2000")
Системная красная волчанка (lupus erithematodes, болезнь Либмана-Сакса) • Распространенность – до 1 на 2000 населения. Женщины 15 -40 лет (75 %) • Этиология – ультрафиолет (инсоляция), инфекция (вирусы, бактерии), медикаменты. HLA DR 2, DR 3 • Патогенез – угнетение Т-супрессоров, антинуклераные аутонтитела (LE-фактор (Ig. G, люпус-фактор), LE-клетки, гематоксилиновые тельца)
 – эритема лица, бабочка,")
ИЗМЕНЕНИЯ ПРИ СИСТЕМНОЙ КРАСНОЙ ВОЛЧАНКЕ • Кожа (до 90 %) – эритема лица, бабочка, дерматит, гиперкератоз, атрофия, язвы • Почки (до 50 %) – гломерулонефрит (волчаночный, неспецифический) • Сердце – эндокардит Либмана-Сакса • Селезенка – гиперплазия, луковичный склероз • Артриты, васкулиты, миозиты, пневмонит, поражение ЖКТ, ЦНС, синдром Рейно

Эритема при СКВ
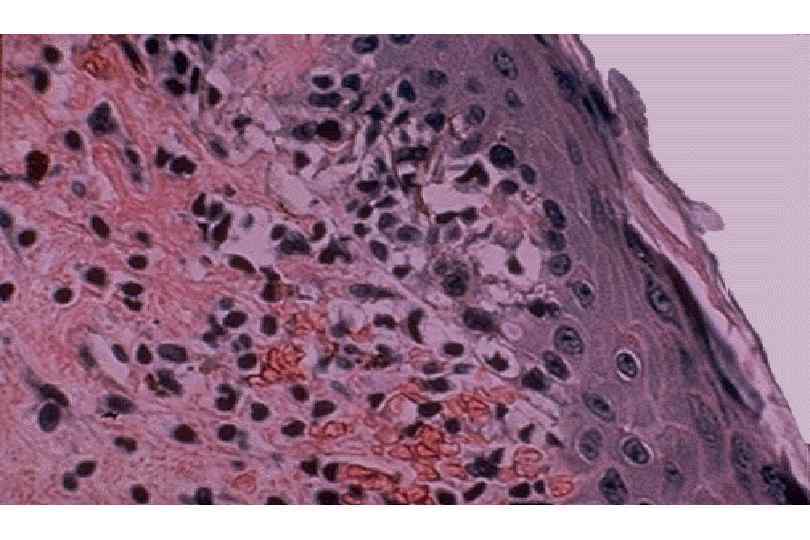
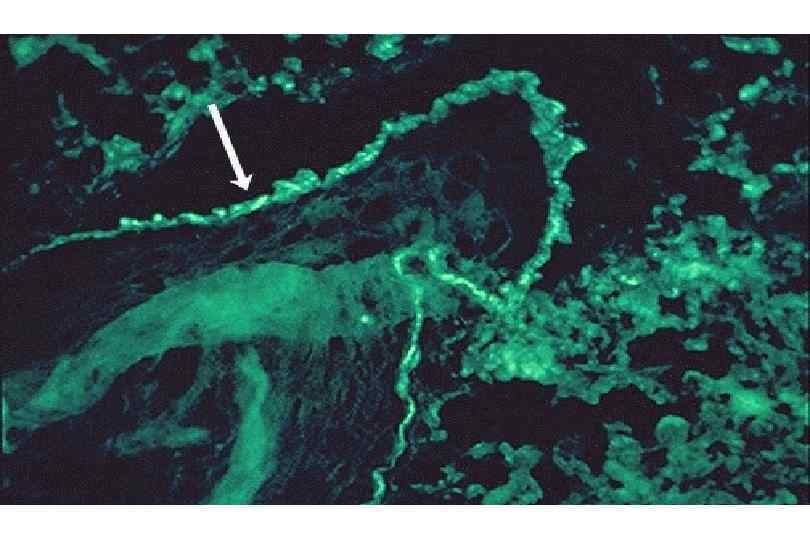
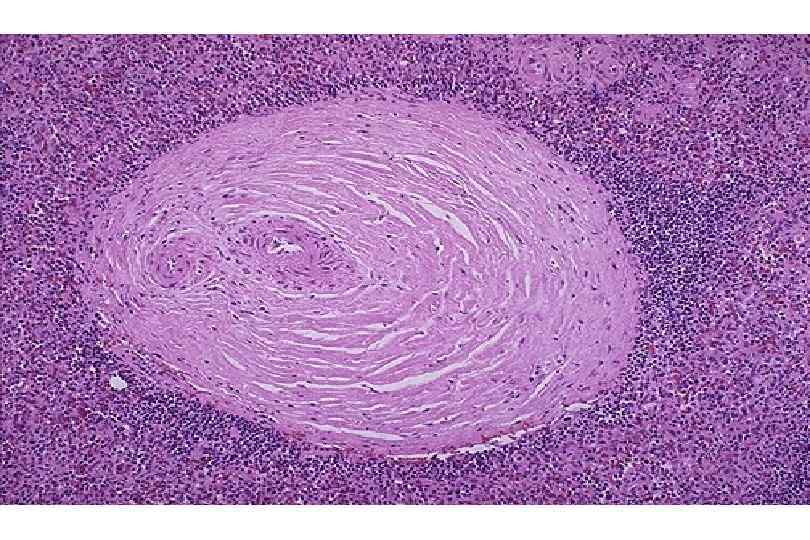
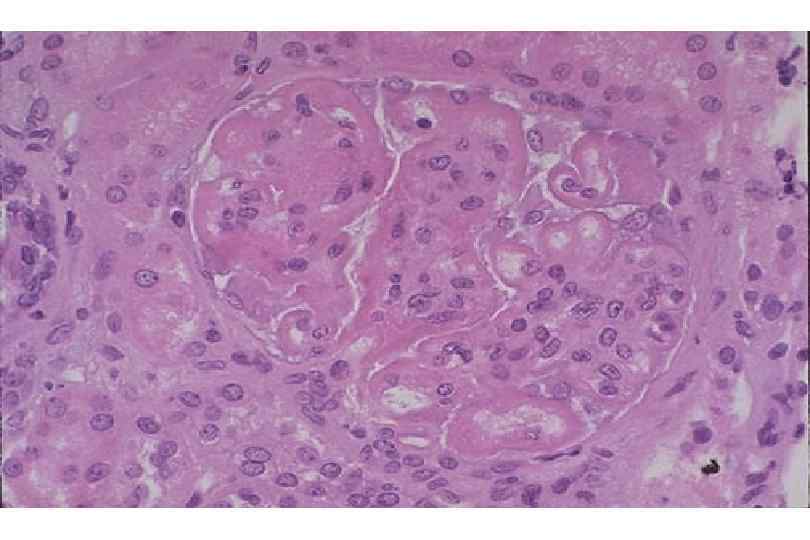

Волчаночный нефрит

Наиболее часто используемые лабораторные тесты: ANA и ANCA

 — нарушения иммунологической реактивности, обусловленные выпадением одного или нескольких компонентов")
Иммунодефициты • Иммунодефициты (ИДС) — нарушения иммунологической реактивности, обусловленные выпадением одного или нескольких компонентов иммунного аппарата или тесно взаимодействующих с ним неспецифических факторов • Единой классификации не существует. По происхождению иммунодефициты делят на первичные и вторичные

КЛАССИФИКАЦИЯ ИММУНОДЕФИЦИТОВ • • • Комбинированные иммунодефициты; Т-клеточные иммунодефициты; Гуморальные иммунодефициты; Дефициты системы фагоцитов; Дефицит системы комплемента.
 дефекты иммунной системы. • гуморальные или")
Первичные иммунодефициты — это врожденные (генетические или эмбриопатии) дефекты иммунной системы. • гуморальные или антительные — с преимущественным поражением системы Влимфоцитов) – Х-сцепленная агаммаглобулинемия (болезнь Брутона) – Гипер-Ig. M синдром • Х-сцепленная • аутосомно-рецессивная – делеция генов тяжелых цепей иммуноглобулинов – дефицит k-цепей – селективный дефицит субклассов Ig. G с или без дефицита Ig. A – дефицит антител с нормальным уровнем иммуноглобулинов – общая вариабельная иммунная недостаточность – дефицит Ig. A

• клеточные – синдром Ди Джоржи – первичный дефицит CD 4 клеток – дефицит CD 7 Т-клеток – дефицит ИЛ-2 – множественная недостаточность цитокинов – дефект передачи сигнала
 – тяжелая комбинированная иммунная")
• комбинированные: – синдром Вискотта-Олдича – атаксия-телеангиоэктазия (синдром Луи-Бар) – тяжелая комбинированная иммунная недостаточность • Х-сцепленная с полом • аутосомно-рециссивная – дефицит аденозиндезаминазы – дефицит пуриннуклеозидфосфорилазы – дефицит молекул II класса МНС (синдром лысых лимфоцитов) – ретикулярная дизгенезия – дефицит CD 3γ или CD 3ε – дефицит СD 8 лимфоцитов • недостаточность системы комплемента
 •")
• дефекты фагоцитоза – наследственные нейтропении • инфантильный летальный агранулоцитоз (болезнь Костмана) • циклическая нейтропения • Семейная доброкачественная нейтропения • дефекты фагоцитарной функции • хроническая гранулематозная болезнь – Х-сцепленная – аутосомно-рецессивная • дефицит адгезии лимфоцитов I типа • дефицит адгезии лейкоцитов 2 типа • дефицит глюкозо-6 -дегидроегназы нейтрофилов • дефицит миелопероксидазы • дефицит вторичных гранул • синдром Швахмана

Лауреаты Нобелевской премии по физиологии и медицине за 2008 год Половину премии, «за открытие вирусов папилломы человека, вызывающих рак шейки матки» , получил Харальд цур Хаузен из Германского центра исследования рака в Гейдельберге. Другую половину премии, присужденную «за открытие вируса иммунодефицита человека» , разделили Франсуаза Барре-Синусси и Люк Монтанье из Института Пастера в Париже.

Стадии инфицирования клеток вирусом СПИД и его репликации • связывание ВИЧ с поверхностью клетки (рецепция); • слияние мембран вируса и клетки и его проникновение внутрь клетки (пенетрация); • высвобождение нуклеоида и геномной РНК; • интеграция геномов клетки и ВИЧ; • активация транскрипции и трансляции после латентного периода; • репликация вируса (морфогенез). • цитопатогенный эффект ВИЧ, высвобождение вирионов из клетки.
 Нуклеопротеины (р7, Vp 2) Gp 120")
Строение ВИЧ-1. РНК Ферменты (РНКаза, обратная транскриптаза, интеграза) Нуклеопротеины (р7, Vp 2) Gp 120 gp 41 Мембрана вириона Латеральные тельца (Vp. X) Коровье белки

ВИЧ-инфекция • Этиология: ретровирус ВИЧ • Пути передачи: половой, парентеральный, трансплацентарный • Тропность: к Т-хелперам (CD-4), ЦНС, макрофаги, эндотелиоциты, эпителиоциты • Изменения: лимфаденопатия, лихорадка, потеря массы тела, диарея, лимфоцитопения, ЦИК • Клинико-морфологические формы: легочная, церебральная, желудочно-кишечная • Фазы: ранняя (острая), средняя (ВИЧ-инфекция), финальная (СПИД) • Оппортунистические инфекции

Оппортунистические инфекции • Оппортунистические инфекции (от лат. opportunus — удобный, выгодный, и лат. infectio — заражение, также англ. opportunity — возможность) — заболевания, вызываемые патогенами (бактерии, вирусы, грибы, простейшие), которые обычно не приводят к болезни у здоровых лиц (с нормальной иммунной системой).

Инфекционные процессы, характерные для СПИД • пневмонии, вызываемые Pheumocystis carinii; • диарея, вызываемая криптоспоридиями, токсоплазмами, лямблиями, амебами; • стронгилоидоз и/или токсоплазмоз головного мозга и легких; • кандидоз полости рта и пищевода; • криптококкоз, диссерминированный или локализованный в центральной нервной системе; • кокцидиомикоз, гистоплазмоз, мукормикоз, аспергиллез различной локализации; • инфекция нетипичными микобактериями различной локализации; • сальмонеллезная бактериемия; • цитомегаловирусная инфекция легких, центральной нервной системы, желудочно-кишечного тракта; • герпетическая инфекция кожи и слизистых оболочек; • инфекция вирусом Эпштейна-Барр; • мультифокальная паповавирусная инфекция с энцефалопатией.
")
Pneumocystis jiroveci (carinii)

Опухоли, связанные со СПИД • саркома Капоши; • неходжкинские лимфомы, локализующиеся преимущественно в головном мозге.
 представляет собой множественные злокачественные новообразования")
Саркома Капоши • Сарко ма Ка поши (ангиосаркома Капоши) представляет собой множественные злокачественные новообразования дермы (кожи). Впервые описана венгерским дерматологом Морицем Капоши и названа его именем.

Саркома Капоши

Саркома Капоши

Амилоидоз • Группа заболеваний, характеризующихся отложением гомогенных эозинофильных нерастворимых белковых масс – амилоида. • Амилоид окрашивается красителем Конго рот в красный цвет и дает двойное лучепреломление и зеленое свечение в поляризационном свете.

Амилоидоз

Амилоидоз

Амилоидоз • Впервые описал Карл Рокитанский – «сальная болезнь» • Название предложил Рудольф Вирхов – «амилоид» – подобный крахмалу

Амилоидоз

Амилоидоз

Амилоидоз • «Большая сальная почка» - отложение амилоида обнаруживается в клубочках, стенках сосудов и строме • «Сальная селезенка» ( «ветчинная селезенка» ) – диффузно в строме и капсуле • «Саговая селезенка» - преимущественно в проекции лимфоидных фолликулов

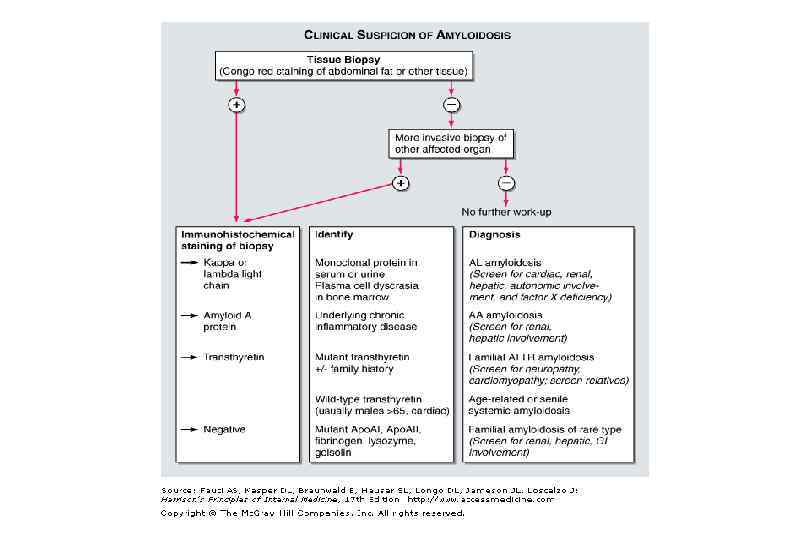
Амилоидоз • Диагноз морфологический • Для постановки диагноза исследуют биопсии: - Подкожно-жировой клетчатки - Слизистой оболочки прямой кишки - Десны - Синовиальной оболочки суставов - Печени - Почки

Амилоидоз

Амилоидоз

Амилоидоз • Актуальность: - Журнал «Amyloid. The Journal of Protein Folding Disoders» выходит с 1994 года - Amyloidosis Support Network http: // www. amyloidosis. org - Банк данных по белкам (The Protein Data Bank) http: //www. rcsb. org/pdb/ - Международные конгрессы и симпозиумы по амилоидозу

Амилоидоз • Патогенез: - Стимул - Растворимый белок предшественник - Образование нерастворимых фибрилл амилоида
")
Амилоидоз • Причины отложения амилоида: - Нарушения в структурной организации белков - Замены (мутации) в аминокислотной последовательности белков

ПРИНЦИПЫ КЛАССИФИКАЦИИ АМИЛОИДОЗА: • ПО ВХОДЯЩЕМУ В СОСТАВ АМИЛОИДА БЕЛКУ – на сегодняшний день выделено более 25 белков • ПО РАСПРОСТРАНЕННОСТИ – системный – местный • ПО ПРОИСХОЖДЕНИЮ – наследственный – приобретенный • ПО КЛИНИЧЕСКИМ ПРОЯВЛЕНИЯМ – с преимущественным поражением тех или иных органов и систем (почек, печени, ЖКТ и т. д. )
 или локальный (L) Синдром или пораженные ткани и")
Название белка Амилоида Белокпредшественник Системный (S) или локальный (L) Синдром или пораженные ткани и органы AL Immunoglobulin light chain S, L AH Immunoglobulin heavy chain S, L ATTR Transthyretin S L? Primary Myeloma-associated Familial Senile systemic Tenosynovium A 2 M 2 -microglobulin AA (Apo)serum AA S L? S Hemodialysis Joints Secondary, reactive AApo. AI Apolipoprotein AI AApo. AII Apolipoprotein AII S L S Familial Aortic Familial AGel Gelsolin S Familial ALys Lysozyme S Familial AFib Fibrinogen -chain S Familial ACys Cystatin C S Familial ABria ABri. PP L, S? Familial dementia, British ADan. PP L Familial dementia, Danish A protein precursor (A PP) L Alzheimer's disease, aging APr. P Prion protein L Spongiform encephalopathies ACal (Pro)calcitonin L C-cell thyroid tumors AIAPP Islet amyloid polypeptide L AANF Atrial natriuretic factor L Islets of Langerhans Insulinomas Cardiac atria APro Prolactin L Alns Insulin L Aging pituitary Prolactinomas Iatrogenic AMed Lactadherin L Senile aortic, media AKer Kerato-epithelin L Cornea; familial A(tbn)b ALac tbnb Lactoferrin L Pindborg tumors L Cornea; familial ADana A

Амилоидоз • Наиболее клинически значимые системные амилоидозы: - Первичный - Вторичный - Наследственный - Диализ-ассоциированный
")
Амилоидоз • Первичный амилоидоз - AL амилоидоз (в состав амилоида входят легкие цепи иммуноглобулинов) • Патогенез: - Неизвестный стимул (канцероген? ) - Моноклональная пролиферация Влимфоцитов - Секреция плазматическими клетками моноклональных L-цепей

Амилоидоз • Первичный амилоидоз (частота встречаемости от 8 на 1 000 до 1 на 100 000 населения в год, остается стабильной в течение 40 лет): - Наблюдается при миеломной болезни - При заболеваниях, сопровождающихся избыточной продукцией моноклональных иммуноглобулинов - Идиопатический

Первичный амилоидоз
 амилоидоз - AА амилоидоз (в состав амилоида входит SAAбелок –")
Амилоидоз • Вторичный (реактивный) амилоидоз - AА амилоидоз (в состав амилоида входит SAAбелок – сывороточный белок предшственник) • Патогенез: - Хроническое воспаление - Активация макрофагов - Продукция интерлейкинов ИЛ 1 и ИЛ 6 - Продукция гепатоцитами белка SAA - Образование АА амилоида
 амилоидоз наблюдается при: - Туберкулез, лепра (до 17% больных) -")
Амилоидоз • Вторичный (реактивный) амилоидоз наблюдается при: - Туберкулез, лепра (до 17% больных) - Анкилозирующий спондилит, ревматоидный артрит, болезнь Крона (0. 5 -13%) - Хронический остеомиелит, хронический абсцесс легкого, бронхоэктазы - Периодическая болезнь

Амилоидоз • Наследственный амилоидоз: - ATTR амилоидоз (транстиретин, преальбумин – плазменный белок связывающий и переносящий тироксин и ретинол) обнаруживается при семейных амилоидных полинейропатиях и старческом семейном амилоидозе

Амилоидоз • Диализ-ассоциированный амилоидоз: - Aβm амилоидоз (β 2 -микроглобулин входит в состав главного комплекса гистосовместимости 1 класса) и определяется при длительном гемодиализе - В мире около 1 млн человек находятся на длительном гемодиализе

Амилоидоз

Амилоидоз

Амилоидоз • Наиболее клинически значимые локальные амилоидозы: - Болезнь Альцгеймера - Сердечный - Инсулярный - Опухолевый
 A-β-2")
Амилоидоз Этиологические формы Предшественник белка амилоида Белок амилоида церебральный амилоидоз трансмембранный гликопротеид (АРР) A-β-2 -протеин изолированный амилоидоз предсердий предсердный натрийуретический фактор (ANF) AANF амилоидоз островков поджелудочной железы при медуллярной карциноме щитовидной железы амилин IAPP кальцитонин ACal

Болезнь Альцгеймера В мире страдает около 12 млн человек

Болезнь Альцгеймера

Болезнь Альцгеймера

Сердечный Амилоидоз

Эндокринный Амилоидоз

Опухолевый Амилоидоз

Опухолевый Амилоидоз

Спасибо за внимание!!!
8 Аутоиммунные болезни Амилоидоз 2012 ред.ppt