Лекция 6-10.ppt
- Количество слайдов: 55



Лекция 17. 04. 2012 Будучи термодинамически неустойчивыми, лиофобные дисперсные системы могут достаточно долго сохранять без изменения исходную дисперсность, т. е. оставаться агрегативно устойчивыми. Это обеспечивается действием различных факторов стабилизации, которые затрудняют протекание процессов, приводящих к разрушению систем. К основным факторам стабилизации относятся: - электростатическое отталкивание диффузных частей двойных электрических слоев (ионно-электростатическая составляющая расклинивающего давления); - «лиофилизация» поверхности за счет адсорбции ПАВ; - образование на поверхности гидрофильных частиц структурированных слоев воды с измененными свойствами по сравнению с объемной жидкостью – повышенной вязкостью (структурная составляющая расклинивающего давления); - структурно-механический барьер (по Ребиндеру).

Расклинивающее давление может быть как положительным, т. е. препятствовать утонению пленки и сближению пластин, так и отрицательным – способствовать утонению пленки и сближению пластин. Расклинивающее давление может иметь различную физикохимическую природу. Соответственно, нужно рассматривать различные составляющие этой термодинамической величины.

Молекулярная составляющая расклинивающего давления обусловлена молекулярным притяжением. Между сближаемыми пластинами действуют силы притяжения, связанные с проявлением дальнодействующих дисперсионных взаимодействий. Так как дисперсионные взаимодействия суммируются по объемам взаимодействующих фаз, результат суммирования дает зависимость энергии взаимодействия (притяжения) в вакууме 2 -х объемов (пластин) в расчете на единицу поверхности от ширины зазора h в виде Umol = - A 11 /12 π h 2 [Дж/м 2], где A 11 – константа Гамакера.

При взаимодействии пластин в конденсированной среде 2, вместо константы A 11 используется сложная константа Гамакера A*, учитывающая взаимодействие молекул фазы 1 и фазы 2: A* = (√A 11 – √A 22)2, Соответственно, энергия притяжения двух объемов фазы 1 в среде 2, выразится как Umol = – A*/12 π h 2 [Дж/м 2], Поскольку имеет место притяжение, Umol < 0.

Umol – это работа, которую необходимо приложить к единице площади пластин, чтобы удалить их с расстояния h на бесконечно большое расстояние в обратимом равновесном процессе. Она равна энергии, которая выделяется при сближении пластин с h = ∞ до данного расстояния h под действием сил молекулярного притяжения. Соответственно, энергия взаимодействия U равна ∆Fпл(h) и, в случае действия только молекулярных сил, Umol < 0 и Π = – d ∆Fпл(h)/ d h = – А*/6 π h 3< 0. Таким образом, при действии только молекулярных сил, происходит самопроизвольное сближение поверхностей пластин и утонение пленки.

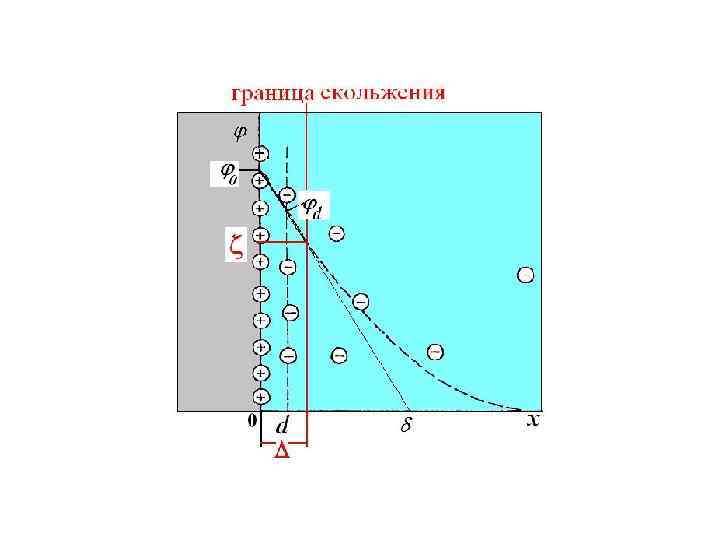
Электростатическая составляющая расклинивающего давления связана с электростатическим отталкиванием диффузных слоев ионов, окружающих частицы дисперсной фазы. При контакте твердых частиц с водной фазой и особенно с раствором электролитов на границе раздела возникает двойной электрический слой (ДЭС). Толщина диффузного слоя δ зависит от концентрации электролита с0, зарядности ионов z и температуры Т: δ ~ √(Т/z 2 с0). При сближении двух поверхностей, на которых имеется ДЭС, при расстоянии h < 2 δ происходит перекрывание диффузных слоев противоионов, сопровождающееся возникновением отталкивания одноименных зарядов (рисунок).
 и потенциал φ(h/2). Произведение ρ(h/2)·φ(h/2) характеризует")
В центре ДЭС h/2 увеличивается плотность зарядов ρ(h/2) и потенциал φ(h/2). Произведение ρ(h/2)·φ(h/2) характеризует плотность электростатической энергии в центре зазора, т. е. работу концентрирования зарядов при сближении поверхностей. Плотность энергии в пленке (Дж/м 3) представляет электростатическую составляющую расклинивающего давления: Πel ~ ρ(h/2)·φ(h/2) [Дж/м 3≡ H/м 2]. собой
 в диффузном слое меняется с расстоянием x по экспоненте φ(x) ~")
Так как φ(x) в диффузном слое меняется с расстоянием x по экспоненте φ(x) ~ exp(–x/δ), Πel также проявляет экспоненциальную зависимость от ширины зазора h: Πel ~ exp(–h/δ). Поскольку электростатическое отталкивание препятствует утонению пленки в зазоре, то Πel > 0. Энергия электростатического взаимодействия Uel соответствует энергии отталкивания (Uel > 0) и также экспоненциально зависит от ширины зазора h: Uel ~ exp(–h/δ). Повышение концентрации с0 приводит к снижению Πel и Uel. Таким образом, электростатическая составляющая Πel и Uel является фактором стабилизации дисперсных систем, т. к. создает препятствие для сближения частиц и их коагуляции.

Структурная составляющая расклинивающего давления связана с образованием на гидрофильных поверхностях структурированных слоев воды. Структура воды в таких тонких пристенных слоях отличается от структуры воды в объеме фазы. Структурированность пристенных слоев воды создает дополнительное сопротивление для сближения частиц и препятствует коагуляции. С ней в значительной степени связаны и иные свойства таких водных пленок – повышенная вязкость и пониженная температура замерзания, играющие большую роль в физических свойствах почв.

Наиболее сильным фактором стабилизации, согласно Ребиндеру, является структурно-механический барьер. Он создается при адсорбции молекул ПАВ, как правило высокомолекулярных, которые способны к образованию структурированного слоя на межфазной границе. К таким веществам относятся глюкозиды, белки, производные целлюлозы (карбоксиметилцеллюлоза) и другие так называемые защитные коллоиды – высокомолекулярные вещества со сложным строением молекул, которые имеют функциональные группы разной гидрофильности в пределах одной молекулы. По отношению к дисперсиям гидрофильных порошков в неполярных жидкостях высокой стабилизирующей способностью обладают многие маслорастворимые ПАВ, способные прочно (химически) адсорбироваться на поверхности гидрофильных частиц. Стабилизированные таким путем лиофобные системы приобретают свойства дисперсий данного стабилизатора, т. е. становятся лиофилизованными. По Ребиндеру, высокую эффективность структурно-механического барьера определяют следующие условия:

1. Наличие повышенной вязкости и механической прочности адсорбционных и межфазных слоев стабилизатора – их способность сопротивляться деформации и разрушению – в сочетании с достаточной подвижностью, обеспечивающей залечивание случайно возникающих дефектов слоя. Для систем с твердыми частицами условием эффективной стабилизации может быть также достаточно высокая прочность закрепления молекул стабилизатора на поверхности частиц, т. е. большая энергия взаимодействия этих молекул с твердой поверхностью (в этих условиях менее существенным становится требование к собственной прочности слоя, обусловливаемой взаимодействием его молекул между собой). 2. Лиофильность наружной части межфазного или адсорбционного слоя, т. е. его родственность дисперсионной среде, обеспечивающая «плавность» перехода от дисперсной фазы к дисперсионной среде.

Коагуляция золей электролитами Устойчивость золей, стабилизированных за счет ионноэлектростатической составляющей расклинивающего давления, оказывается весьма чувствительна к добавкам электролитов. По мере увеличения концентрации электролита заметная коагуляция проявляется только выше некоторой критической концентрации ск, называемой порогом коагуляции. Величина, обратная порогу коагуляции получила название коагулирующей способности γ = 1/ ск.
 скорость")
dn/dt Ck C При увеличении концентрации электролита выше порога коагуляции (с > ск) скорость коагуляции dn /dt, т. е. изменение числа частиц в единицу времени, сначала нарастает (участок I рисунка) – это область «медленной коагуляции» , а затем перестает зависеть от концентрации электролита (участок II) – область «быстрой коагуляции» . Области быстрой коагуляции соответствует полная дестабилизация дисперсной системы.

Существуют эмпирические правила, связывающие явления коагуляции с характером коагулирующих ионов и электроповерхностными свойствами золей (ζ-потенциалом). Согласно правилу Шульце-Гарди, коагулирующее действие электролитов определяется зарядом z (валентностью) противоионов: отношение порогов коагуляции одно-, двух- и трехвалентных ионов приближенно равно 1 : 0, 016 : 0, 0015; соответственно, их коагулирующие способности γ относятся как 1 : 60 : 700, т. е. коагулирующая способность возрастает приблизительно пропорционально шестой степени заряда ионов (z 6). Заметная коагуляция начинается, когда под воздействием электролита ζ-потенциал снижается до некоторого критического значения (ζк), составляющего примерно 30 м. В. Это значение достигается при концентрации электролита, соответствующей порогу коагуляции. При вхождении в область быстрой коагуляции ζпотенциал падает до очень малых значений.

Электролиты, содержащие крупные поливалентные катионы (Al 3+ , Fe 3+, Th 4+ и т. п. ) или анионы, способные перезарядить коллоидные частицы и сменить знак -потенциала, по мере их добавления к золю могут вызвать сначала коагуляцию, затем пептизацию (самопроизвольное диспергирование коагулята), а при дальнейшем увеличении концентрации – повторную коагуляцию. В этом случае наблюдаются так называемые "неправильные ряды" – чередование зон устойчивости и коагуляции: золь проходит две зоны устойчивости и две зоны коагуляции. Этим двум зонам соответствуют два различных порога коагуляции (ck 1 и ck 2 ) и одинаковые по величине, но противоположные по знаку критические значения ‑потенциала (рисунок).

-z -zкр 0 C zкр +z Ск 1 Ск 2 Области концентраций, для которых -потенциал имеет значения (по абсолютной величине) больше критического | | > | kр |, отвечают зонам устойчивости, а области, где | | < | kр | – зонам коагуляции.

Основы теории ДЛФО Теория коагулирующего действия электролитов развита Б. В. Дерягиным совместно с Л. Д. Ландау в 1941 году и несколько позже независимо в работах голландских ученых Э. Фервея и Я. Овербека (теория ДЛФО). В основе ее лежит рассмотрение коагуляции как результата совместного действия двух противоположных факторов: ван-дер-ваальсового притяжения и электростатического отталкивания диффузных слоев противоионов, окружающих коллоидные частицы. Энергия взаимодействия частиц U определяется суммой двух величин U = Umol + Uel, где Umol – энергия молекулярного притяжения (Umol< 0), а Uel – энергия электростатического отталкивания (Uel> 0).

Эти составляющие суммарной энергии взаимодействия U по разному зависят от расстояния h между частицами: Umol ~ 1/h 2, Uel ~ exp(-h/ δ), где δ – толщина диффузного слоя противоионов, зависящая от концентрации электролита в объеме с0 δ ~ .
 пока не перекрываются диффузные слои противоионов (Uel")
На больших расстояниях ( h >>2δ ) пока не перекрываются диффузные слои противоионов (Uel ≈ 0) между частицами действуют только силы молекулярного притяжения и UΣ = Umol< 0. После сближения до расстояний h < 2δ между частицами возникают силы отталкивания, что приводит к уменьшению по абсолютной величине UΣ. По мере дальнейшего сближения частиц, степень перекрывания диффузных слоев увеличивается и на некоторых средних расстояниях Uel становится преобладающей над Umol, что приводит к возникновению энергетического барьера UΣ > 0. При сближении до очень малых расстояний, несмотря на сильное перекрытие диффузных слоев, молекулярное притяжение Umol нарастает сильнее, чем Uel, и UΣ сначала уменьшается, а затем переходит в отрицательную область, что соответствует преобладанию Umol над Uel. На кривой зависимости UΣ(h) появляется максимум. Высота и положение максимума зависят от концентрации электролита с0 в объеме среды, поскольку от этого зависит толщина и заряд диффузного слоя и, соответственно, электростатическое отталкивание Uel.
 могут")
В результате неодинаковой зависимости Umol и Uel от расстояния h на зависимости UΣ(h) могут обнаруживаться два минимума (на малых и больших расстояниях) и один максимум (на средних расстояниях) – рисунок. U Uel 2 d Umol h Umol

Добавки электролита прежде всего влияют на электростатические силы отталкивания. При добавлении электролита происходит уменьшение толщины диффузного ионного слоя; на суммарной зависимости U(h) уменьшается величина потенциального барьера и происходит сдвиг положения максимума в сторону меньших толщин (кривая 2 на рис. 2). При определенной концентрации электролита барьер полностью исчезает, что соответствует полной потере системой агрегативной устойчивости (кривая 3 рис. 2). Происходит коагуляция. повышение концентрации электролита

Флокуляция Близким по внешним проявлениям к процессу коагуляции является флокуляция, наблюдаемая при добавлении к золям некоторых высокомолекулярных ПАВ – флокулянтов – в очень малых количествах (обычно не выше 0, 01%). Хорошие флокулирующие свойства проявляют полиэлектролиты – водорастворимые высокомолекулярные ПАВ, содержащие в своей цепи множество функциональных групп, которые при диссоциации в водной среде образуют большие полиионы. При флокуляции образуются более рыхлые, чем при коагуляции, агрегаты, в которых частицы находятся на значительных расстояниях и связаны между собой мостиками из макромолекул полимеров. При малых концентрациях молекулы таких ПАВ оказываются достаточно «развернутыми» и могут закрепляться сразу на нескольких частицах дисперсной фазы, связывая их (особенно прочно при хемосорбции) в единый рыхлый агрегат (рисунок). При высоких концентрациях полимерного ПАВ, когда адсорбция на поверхности частиц велика и сопровождается образованием плотного лиофилизующего адсорбционного слоя, тот же полимер будет выступать в роли стабилизатора дисперсии, действующего по механизму структурномеханического барьера.

флокуляция стабилизация

Флокуляция с помощью полимеров нашла широкое применение во многих прикладных областях: в водоочистке для ускорения осаждения частиц (и особенно досаждения мелких фракций), в агротехнике для управления фильтрационными и структурно-механическими свойствами почв, в том числе для предотвращения ветровой эрозии (что особенно актуально после чернобыльской катастрофы), в инженерной геологии для закрепления грунтов, в бумажной промышленности для регулирования связности целлюлозных волокон на разных этапах переработки бумажной пульпы, фармации – для выделения активных веществ в определенной форме и т. д. В качестве флокулянтов водных дисперсий используются различные полиэлектролиты – полиакриламиды, полиэтиленимины и другие.

Структурированные дисперсные системы Нарушение агрегативной устойчивости дисперсных систем сопровождается переходом свободнодисперсной системы в связнодисперсную и в определенных условиях может приводить к развитию во всем объеме системы пространственных сеток – дисперсных структур. В таких структурированных системах силы сцепления в контактах между частицами достаточно велики, чтобы противостоять не только тепловому движению, но и внешним воздействиям. В результате система приобретает комплекс новых – структурно-механических (реологических) свойств, характеризующих ее сопротивление деформации и разрушению.

L Традиционная реология = При ламинарном сдвиговом течении жидкости между двумя плоскопараллельными пластинами, верхняя из которых под действием силы F движется с постоянной скоростью v, а нижняя неподвижна, слои перемещаются с разными скоростями – от максимальной у верхней пластины до нуля – у нижней. В зазоре реализуется постоянный градиент скорости. Под действием силы F в ходе перемещения верхней пластины на расстояние L в системе возникает напряжение сдвига (касательное или тангенциальное напряжение): . Единицей напряжения является Паскаль (Па = Н/м 2). Градиент скорости равен dv/dh. Вязкость h = / - закон Ньютона.
 сдвиговом течении W~ (cкорость сдвига)")
Традиционная реология: Мы будем говорить только о стационарном (непрерывном) сдвиговом течении W~ (cкорость сдвига) M~ (напряжение сдвига) Вязкость: h = / [Па. с] - закон Ньютона

Два типичных способа отображения реологического поведения дисперсных и полимерных систем: Кривые течения Зависимости вязкости от скорости сдвига lg h lg lg При низких скоростях реализуется постоянная (наибольшая ньютоновская) вязкость

Согласно классическим работам П. А. Ребиндера аномалия вязкости в структурированных системах достигает 7 -8 порядков! Причина: «…механическое разрушение пространственной сетки – беспорядочной структуры…» . Подразумевается, однородное! Так ли это? Можно ли визуализировать сдвиговое течение суспензий Naмонтмориллонита?
 1. Регулируемый источник тока 2. Зеркало 3.")
Схема прибора для плоскопараллельного сжатия (“squeezing flow”) 1. Регулируемый источник тока 2. Зеркало 3. Матовое стекло 4. Поляроид 1 5. Постоянный магнит 6. Электромагнит 7. Предметный стол 8. Верхняя и нижняя плоскости измерительной ячейки 9. Исследуемый образец 10. Объектив микроскопа 11. Поляроид 2

Схема метода плоскопараллельного сжатия а б При сжатии капли возможны два способа реологических испытаний: с постоянным объемом (а) и постоянной площадью (б).

Течение суспензии Halloysite + H 2 O в условиях плоскопараллельного сжатия
 пр r Роль гидростатического давления?")
Суспензия Cloisite Na+ + ПЭГ(промежуточная фаза сжатия) пр r Роль гидростатического давления?

Структурообразование в дисперсных системах следует рассматривать в связи с другим фундаментальным явлением – устойчивостью дисперсных систем. Результатом потери устойчивости могут быть коагуляция и седиментация с последующей коалесценцией, вплоть до полного разделения исходной системы на макроскопические фазы: происходит «гибель» дисперсной системы. Однако утрата частицами дисперсной фазы прежней подвижности, обусловленная сцеплением частиц, может иметь следствием формирование пространственной сетки. В итоге возникает структура с определенными механическими свойствами, и дисперсная система получает новую «жизнь» – она становится материалом.

Наиболее широко используемая механическая характеристика таких структур – их прочность, т. е. способность сопротивляться разрушению под действием механических напряжений. Как и другие механические свойства дисперсных структур (упругость, пластичность, повышенная вязкость), прочность зависит не столько от свойств частиц, образующих структуру, сколько от величины сил сцепления частиц и степени «развитости» структуры.

В современном физическом языке существует термин «перколяция» , соответствующий формированию в дисперсной системе непрерывного кластера. Перколяционный порог, т. е. объемная концентрация образования такого кластера существенно зависит от степени анизометрии частиц. Так, для сферических частиц сажи в полимерах она составляет 10 -15%, а для углеродных нанотрубок – менее 1 %.

Итак, в результате благоприятных броуновских соударений частицы, сцепившись по углам или ребрам, способны образовать структурную сетку – достаточно жесткий каркас. Для широкого круга дисперсных структур в первом приближении Рс ≈ χр1, где Рс – прочность дисперсной структуры [Па], χ – число контактов на единицу поверхности разрушения [м-2], р1 – средняя прочность индивидуальных контактов [Н]. При этом под прочностью индивидуального контакта подразумевается сила, необходимая для его разрушения (разъединения частиц). Механические свойства дисперсных структур можно варьировать в очень широких пределах с помощью направленных физико-химических воздействий на систему в процессе ее формирования.

В зависимости от типа контакта, т. е. от характера сил, обуславливающих сцепление частиц, различают два типа дисперсных структур: структуры с контактами коагуляционного типа и структуры с фазовыми контактами. В коагуляционных структурах взаимодействие частиц в контактах ограничивается их "соприкосновением" через сохраняющиеся равновесные прослойки дисперсионной среды (а) или непосредственным (б). Для таких контактов и структур в целом характерна механическая обратимость – способность к самопроизвольному восстановлению после механического разрушения (тиксотропия). Возможность коагуляции в системе определяется конкуренцией между сцеплением частиц и их участием в тепловом движении. Дисперсная система будет термодинамически устойчива относительно коагуляции, если работа диспергирования WД оказывается меньше, чем выигрыш свободной энергии системы из-за увеличения энтропии ΔS при включении частиц в броуновское движение.
z– число контактов в агрегате,")
Так как (uk – энергия связи частиц в контакте; (½)z– число контактов в агрегате, z – координационное число), а ΔS×Т = Nβk. T , где β определяется отношением концентрации частиц дисперсной фазы в агрегированном na и пептизированном nп состояниях ( β ≈ ln (na / nп) ), из условия Wд < ΔS×Т следует, что при uk< βk. T/(½)z дисперсная система будет термодинамически устойчива относительно коагуляции. Наоборот, при uk> βk. T/(½)z коагуляция термодинамически выгодна. Тогда в качестве критерия устойчивости системы относительно коагуляции можно использовать величину энергии взаимодействия частиц в контакте uk = uk* , при которой в системе устанавливается равновесие коагуляция ↔ пептизация: uk = uk* = βk. T/(½)z Для разбавленных золей, образующих рыхлые коагуляты (β ≈ 10÷ 20, z ≈ 2÷ 3), критическое значение uk* составляет 10÷ 15 k. T.

В этом рассмотрении величина uk соответствует глубине первичного потенциального минимума на кривой зависимости избыточной свободной энергии ΔF (h), т. е. энергии взаимодействия частиц на равновесном расстоянии h 0 в коагуляционном контакте: где A* - сложная константа Гамакера, r – радиус кривизны поверхности частиц в месте их соприкосновения. Тогда сила сцепления частиц (прочность единичного коагуляционного контакта), обусловленная силами Ван-дер. Ваальсова притяжения, запишется как: При А* ≈ 10 -19 Дж и h 0 ≈ 1 нм между частицами радиуса 1 мкм действует сила около 10 -7 Н, что вполне поддается непосредственным измерениям.

При лиофилизации системы (в результате адсорбции ПАВ на границе раздела твердая частица – дисперсионная среда или при замене жидкой среды на более родственную твердой фазе) межфазная энергия σ и сложная константа Гамакера А* могут значительно (на 2 – 3 порядка) снижаться. В высококонцентрированной системе это приводит к потере прочности или пластифицированию системы. В отсутствие жидкой прослойки среды в зазоре между частицами (при прорыве адсорбционно-сольватной оболочки) достигается непосредственное «точечное» соприкосновение частиц, т. е. площадь контакта соответствует одной или нескольким атомным ячейкам. В этом случае наряду с ван-дер-ваальсовыми, действуют также и близкодействующие валентные силы. Порядок их величины (без учета специфики химических связей) можно оценить как , где n – число валентных связей на один контакт, e – заряд электрона, b – характерное межатомное расстояние. Как и в предыдущем случае, сила взаимодействия в точечном контакте составляет около 10 -7 Н.

Однако даже в том случае, когда взаимодействие частиц обусловлено только дисперсионными силами, рассчитать глубину минимума весьма проблематично, т. к. точные значения констант Гамакера частиц и среды известны для очень ограниченного круга объектов. Поэтому для обоснования критерия устойчивости необходимо получить непосредственные данные об энергии взаимодействия модельных частиц в первичном потенциальном минимуме и сопоставить их с поведением реальной коллоидной системы с такими же частицами. Путь к решению этой проблемы открывает теория Дерягина, устанавливающая связь между силой притяжения частиц дисперсной фазы, разделенных зазором h (т. е. прочностью контакта p 1), кривизной частиц и величиной U(h) – энергией притяжения двух фаз, разделенных плоским зазором той же толщины: , где k – геометрический параметр, определяемый кривизной контактирующих поверхностей.

Для двух сферических частиц с радиусами r 1 и r 2 это выражение принимает вид: p 1 = –p r¯ U(h) , где Теория носит чисто термодинамический характер, так как не делается никаких предположений относительно характера сил, обусловливающих сцепление.
 связана с межфазным натяжением на границе раздела частица дисперсной фазы (S) –")
Величина U(h) связана с межфазным натяжением на границе раздела частица дисперсной фазы (S) – дисперсионная среда (L) как: U(h) = 2σSL – ΔσПЛ Величину ΔσПЛ называют избыточным натяжением пленки, связанным с совершением работы d. W = –П(h)dh при утонении пленки (П– расклинивающее давление). При контакте частиц в жидкой среде величина ΔσПЛ – это работа образования симметричной пленки толщиной h. Очевидно, что ΔσПЛ ≥ 0. При непосредственном контакте жидких частиц, когда разрыв пленки приводит к их слиянию и полному исчезновению границы раздела между ними, ΔσПЛ = 0.

Таким образом, измеряя силы сцепления p 1 между частицами известного радиуса, приведенными в контакт в жидкой среде, можно определить удельную энергию взаимодействия U – величину, инвариантную относительно размеров частиц и зависящую, только от свойств межфазной границы. Из соотношения uk = p 1× h 0 можно оценить глубину первичного минимума и, сопоставив с его критическим значением uk, оценить устойчивость коллоидных дисперсий с такими же по природе частицами.
,")
Принципиально иным типом контактов, по сравнению с коагуляционными, являются фазовые контакты (в и г), в которых сцепление между частицами обусловлено близкодействующими силами когезии. Структуры с фазовыми контактами П. А. Ребиндер называл конденсационно-кристаллизационными; они отличаются высокой прочностью, необратимым характером разрушения, отсутствием тиксотропных свойств.
 к")
Во всех этих случаях имеет место переход от простого соприкосновения частиц (коагуляционных контактов) к возникновению когезионного взаимодействия. Такой переход связан с протеканием в дисперсной системе необратимых процессов (спекание, пластическая деформация, кристаллизация). Характер этих процессов, приводящих к возникновению фазовых контактов, делает их, в отличие от коагуляционных, механически необратимыми, т. к. разрушение структуры с фазовыми контактами не возвращает систему в исходное состояние. Механизм образования фазовых контактов может быть различным, а результат один – переход от соприкосновения к когезии на значительной площади.

В фазовых контактах когезионное взаимодействие реализуется на площади, заметно превышающей площадь элементарной ячейки, а сама контактная поверхность подобна границе зерна в поликристаллическом материале. Если размер контакта меньше расстояния между дефектами в твердом теле, то его прочность приближается к теоретической прочности идеального твердого тела (pid). Таким образом, максимальная оценка силы сцепления в таких контактах дает величину где s – площадь контакта. p 1 = pid∙s, Структуры с фазовыми контактами могут возникать в процессах спекания порошковых материалов при значительном повышении температуры или в условиях «холодной сварки» , когда истинное сцепление за счет близкодействующих сил осуществляется в результате пластической деформации контактирующих частиц при напряжениях выше предела текучести.

Срастание частиц происходит в результате растворения материала в напряженных контактах и его кристаллизации в зазорах между частицами (жидкофазное спекание). Под действием внешних напряжений в присутствии жидкой фазы наблюдается заметная деформация (компакция) порошка. Этот процесс лежит в основе такого нежелательного явления, как слеживание сыпучих материалов.

Когда гидратация исходного вяжущего вещества происходит в достаточно концентрированных суспензиях и возникающие кристаллики бигидратных образований, соприкасаясь друг с другом, могут удерживаться в фиксированном положении, кристаллизация сопровождается срастанием отдельных кристалликов между собой с образованием фазовых кристаллизационных контактов в единую камневидную структуру.
 Сферическая пена Полиэдрическая пена Газовая фаза составляет до 95% пены.")
Газовая дисперсная фаза (пены) Сферическая пена Полиэдрическая пена Газовая фаза составляет до 95% пены. Оставшиеся 5% приходятся на жидкую (вода) или твердую фазу (полимеры). Пена всегда находится в термодинамически неравновесном состоянии. В отсутствии ПАВ пены, как и другие дисперсные системы, неустойчивы, поскольку минимуму свободной энергии отвечает макроскопический распад на фазы.

При формировании газовых пузырьков в жидкости они сохраняют сферическую форму только при малом объемном наполнении. В дальнейшем упаковка становится более плотной (полиэдрической), в которой пузырьки разделены тонкими пленками жидкости. Три пузырька с пятиугольными гранями и углами при вершинах, равными 120 о, сходятся в одном узле. Ребрами пенной ячейки являются заполненные жидкостью каналы Плато. Со временем жидкость вытекает из пленок в каналы. Пленки становятся такими тонкими, что границы начинают взаимодействовать друг с другом. Это начало гибели пены и только добавки ПАВ позволяют продлить ее жизнь. * *

Фазовые диаграммы; золь-гель переход Существует два основных вида фазовых равновесий: аморфное и кристаллическое/жидкокристаллическое. В первом случае состояние системы описывается бинодалью (а), во втором – линией ликвидуса (б). Т Т раствор (золь? ) НКТС ВКТС а) б) распад на фазы (гель ? ) А Б В заштрихованной области система двухфазна, за пределами ее – однофазна. В зависимости от температурной зависимости термодинамического потенциала (для полимеров параметра Флори-Хаггинса) системы могут быть с верхней (ВКТС) или нижней температурой смешения (НКТС). Под линией ликвидуса – гель, выше – раствор (золь).
 ПАВ В заштрихованной")
Для коллоидных систем часто используют тройные диаграммы состояния (в виде треугольника) ПАВ В заштрихованной области (при определенном соотношении компонентов) система распадается на аморфные фазы. масло вода ПАВ г м с к Как правило, диаграммы состояния коллоидных систем более сложные и могут состоять из нескольких бинодалей или линий ликвидуса, а также промежуточных линий. а вода масло
Лекция 6-10.ppt