Лейкозы.pptx
- Количество слайдов: 160


 с курсом гериатрии и физиотерапии ГБУ")
ЛЕЙКОЗЫ Асс. кафедры общей врачебной практики (семейной медицины) с курсом гериатрии и физиотерапии ГБУ Рост. ГМУ, к. м. н. Шнюкова Т. В.

Гемобластозы Гемобластозами называются опухоли, возникающие из кроветворных клеток. В зависимости от первичной локализации патологического процесса гемобластозы подразделяются на лейкозы и гематосаркомы (злокачественные лимфомы).

Лейкозы – опухоли из гемопоэтических клеток, первично поражающие костный мозг. Лейкозы делятся на острые и хронические. При острых лейкозах опухолевую массу составляют молодые, недифференцированные или малодифференцированные (бластные) клетки. В группу хронических лейкозов входят заболевания, морфологический субстрат которых представлен зрелыми клетками.
, чаще")
Злокачественные лимфомы При злокачественных лимфомах опухолевый процесс первично локализуется вне костного мозга (экстрамедуллярно), чаще всего в лимфатических узлах. В соответствии с гистологической картиной злокачественные лимфомы разделяются на лимфогранулематоз и нелимфогранулематозные (неходжскинские) лимфомы. При прогрессировании лимфом патологические клетки метастазируют в костный мозг и появляются в периферической крови. На этой стадии лейкемизации клинические различия между лейкозами и лимфомами практически исчезают.


Распространенность По уровню заболеваемости гемобластозы занимают 5 -6 место среди злокачественных новообразований, что составляет 5 -6 на 100 тысяч населения. Соотношение между лейкозами и лимфомами 1: 1. При всех формах гемобластозов заболеваемость мужчин выше, чем женщин. Пики заболеваемости: 2 -4 года, 10 -12 лет, старше 50 (чаще 60 -69 лет).
. Единой причины гемобластозов не существует. Вирусно-генетическая концепция (ведущая). Подтверждается: I) у животных.")
Этиология (1). Единой причины гемобластозов не существует. Вирусно-генетическая концепция (ведущая). Подтверждается: I) у животных. а) У большинства животных, способных болеть лейкозом (птицы, грызуны, куры, свиньи, крупный рогатый скот, шимпанзе), выделен вирус, который называется онкорновирус (содержит РНК). б) Считается, что вирус у животных передается по наследству по вертикали. в) Повышение заболеваемости у животных вызвало повышение заболеваемости у обслуживающего персонала.
. II) У человека. До 15 вирусов предлагаются как этиологический фактор гемобластозов. Из")
Этиология (2). II) У человека. До 15 вирусов предлагаются как этиологический фактор гемобластозов. Из крови людей, больных гемобластозами, выделен ретровирус типа С, сходный с онкогенными вирусами обезьян. В 80 -х годах выделен вирус от больного лимфомой Бёркита. В 1982 году выделен ретровирус от больного Т-клеточным лейкозом и назван человеческим Т-клеточным вирусом (НТЛВ-1). В настоящее время этот вирус и антитела к нему выделяют от больных лейкозом, мед. персонала, изредка у интактных лиц, у больных гемофилией, получавших гемотрансфузии. НТЛВ-2 недавно выделен у больного волосато-клеточным лейкозом. Вирус Н 4, близкий вирусу лейкоза, выделен при СПИДе. Считается, что клетки позвоночных содержат наследственное вещество онкорновируса. Присутствие этого вещества в половых клетках обеспечивает вертикальную передачу от родителей к детям. Это позволяет ретроспективно говорить о том, что лейкоз, как и все онкозаболевания, – это заболевание с генетическим детерминированием.
. Вирус поддерживается в латентном состоянии при помощи системы иммунитета, генов супрессоров. Срыв")
Этиология (3). Вирус поддерживается в латентном состоянии при помощи системы иммунитета, генов супрессоров. Срыв в системе иммунологической толерантности, который может наступить под воздействием различных лейкогенных факторов, означает начало заболевания. Однако не всякое воздействие вируса сопровождается образованием лейкозного клона: клон должен иметь свои генетические стигмы. Вирус, содержащий РНК, с помощью обратной транскриптазы внедряется в матрицу ДНК клеток хозяина. В данном случае этими шоковыми клетками являются стволовые клетки-предшественники миело- или лимфопоэза. В результате внедрения вируса в эти клетки они получают новую генетическую информацию, вызывающую непрерывную пролиферацию клетки без дифференцировки.

Против вирусной теории в этиологии выступают факторы, которые в настоящее время играют все меньшее значение: а) установлен факт неконтагиозности лейкоза; б) редкость развития у детей, матери которых во время беременности болели лейкозом; в) отсутствие передачи заболевания при переливании крови от больного с лейкозом.

Концепция радиационной и химической теории В Японии у людей, подвергшихся атомной бомбардировке, существенно возросла частота хронического миелолейкоза, а в последующем – острого миелобластного лейкоза. Значительно чаще встречаются лейкозы и лимфомы у людей, получавших лучевую терапию по поводу различных заболеваний, а также после применения радиоактивных изотопов с лечебной целью. Риск заболевания лейкозами увеличен у людей, имеющих длительный контакт с бензолами, летучими органическими растворителями. Частота лейкозов повышается у лиц, получавших лечение цитостатиками и особенно при сочетанном применении цитостатиков и лучевой терапии. Так, у больных лимфогранулематозом, которые лечились комбинацией цитостатиков МОРР и облучением, частота лейкозов достигала 6 -10%.
. Зарегистрирован")
Предрасполагающие факторы: 1. физические; ионизирующая радиация, физические рентгенологические исследования (чаще – миелобластный лейкоз). Зарегистрирован дозозависимый лейкогенный заряд при облучении позвоночника больных спондиллезом. 2. Химические: а) производственные – Химические полициклические лаки, краски, бензол, пестициды, тетраэтилсвинец, б) лекарственные – бутадион, левомицетин, метилурацил, длительное применение солей золота, висмута. 3. Биологические (в том числе инфекционные): вирусный гепатит В, сальмонеллез, дизентерия, туберкулез; беременность, роды, аборты; хирургические вмешательства; стресс.

Патогенез. Клональная теория 1. онкорновирус внедряется при помощи обратной транскриптазы в матрицу ДНК-содержащих стволовых клеток. Происходит изменение генома стволовых клеток, изменение интимных отношений между ДНК и РНК с повреждением хромосомного аппарата клеток, с гибридизацией вирусных генов с клеточными генами и образованием канцерогенных генов. Стволовая клетка производит дочерние клетки, т. е. клоны клеток. В начале опухоль носит моноклональный характер, т. е. клетки по своему геному родственны стволовой клетке (I – моноклоновая фаза патогенеза). С каждым последующим клоном увеличивается генетическая нестабильность и постепенно в первоначальном опухолевом клоне появляются новые мутантные клоны – опухоль приобретает поликлоновый характер (II – поликлоновая фаза патогенеза). При этом обеспечивается преимущественное размножение наиболее злокачественных клонов, что приводит к прогрессирующему угнетению нормального гемопоэза, метастазированию гемобластозов вне органов кроветворения, появлению резистентности к ранее эффективному лечению.

Механизмы вытеснения нормальных ростков кроветворения опухолевыми клетками: Токсины, подавляющие тот или иной росток кроветворения; Активация фиброза в костном мозге; Борьба за продукты питания; Механическое вытеснение; Аутоиммунное воздействие (характерно преимущественно для хронического лимфолейкоза).

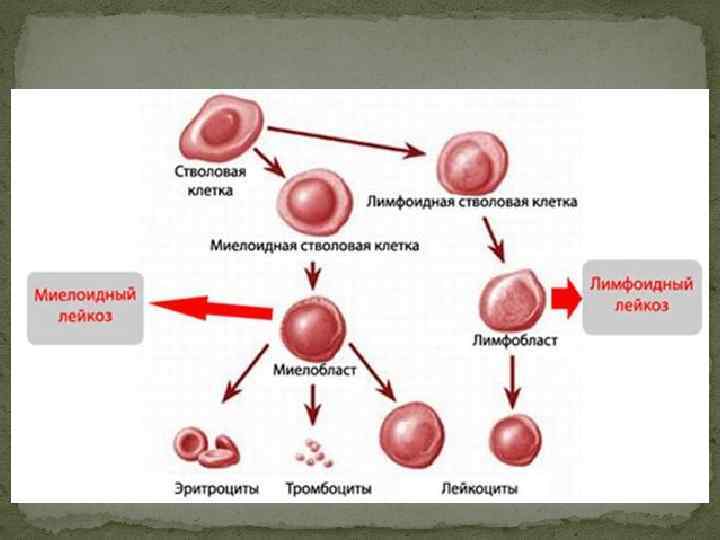
ОСТРЫЙ ЛЕЙКОЗ Злокачественное опухолевое заболевание кроветворной системы, начинающееся с костного мозга и характеризующееся накоплением недифференцированных (бластных) клеток с подавлением нормальных ростков костного мозга. В прошлом лейкозы называли лейкемиями (белокровием) по одному важному, но не обязательному признаку – появлению в крови опухолевых лейкоцитов.
. 1. Острый лимфобластный лейкоз. 60% (85% у детей,")
Классификация французско-американобританской рабочей группы (FAB-классификация, 1980). 1. Острый лимфобластный лейкоз. 60% (85% у детей, 15% у взрослых). На основании иммунофенотипирования бластных клеток подразделяется: а) Т-форма – 15 -25%, мембраны бластов имеют маркеры Т-клеток; б) В-форма – 3 -5%, мембраны бластов имеют маркеры В-клеток; в) общий острый лимфобластный лейкоз – 60%, бласты экспрессируют общий антиген (callантиген), но не имеют других антигенов, свойственных Т- или В-клеточным линиям; г) нуль-острый лимфобластный лейкоз – 10%, лейкозные клетки не имеют ни В, ни Тмаркеров и не экспрессируют общего антигена (call-антигена), выявляется лишь общий для лимфоидной популяции маркер – пан-Т-антиген; д) гибридные формы острого лимфобластного лейкоза – 1 -7%, в лейкозных клетках одновременно присутствуют маркеры лимфоидной и миелоидной направленности. 2. Острые нелимфобластные лейкозы: а) миелобластный: М 1 – острый миелобластный лейкоз без признаков вызревания клеток (20% случаев); М 2 - острый миелобластный лейкоз с признаками вызревания клеток (30% случаев). б) промиелоцитарный – М 3 (8%). в) миеломонобластный – М 4 (28%). г) монобластный – М 5 а – без созревания клеток, М 5 б – с частичным созреванием клеток (10%). д) эритробластный – М 6 (4%). е) мегакариоцитарный – М 7. ж) острый недифференцированный лейкоз – М 0.

Диагностика острых лейкозов строится на данных цитологического исследования крови и костного мозга, обнаруживающих высокий процент властных клеток. На ранних этапах их в крови обычно нет, но выражена цитопения. Поэтому при цитопении, даже касающейся одного ростка, необходима пункция костного мозга, которую можно делать амбулаторно. Диагноз острого лейкоза ставят при наличии в костном мозге не менее 30% бластов. При наличии в костном мозге бластов менее 30% речь идет о малопроцентном лейкозе – варианте миелодиспластического синдрома (рефрактерная к лечению анемия в сочетании с различными цитопениями и дисгемопоэзом. В костном мозге при этой форме, как правило, процент бластов меньше, чем в крови. Малопроцентная форма острого лейкоза практически не поддается лечению, возможно выполнение аллогенной трансплантации костного мозга или паллиативная терапия). Для верификации типа лейкоза проводят морфологию бластов и цитохимический анализ бластов. Реакция на гликоген (ШИК-реакция): лимфобласты красятся гранулами, нелимфобласты (и нейтрофилы) – диффузно. Реакции на миелопероксидазу (PAS-реакция), эстеразу, нафтилэстеразу, пероксидазу, фосфолипиды отрицательны при лимфобластном варианте, положительны при прочих видах острых лейкозов.

В последнее время для точной диагностики ОЛ дополнительно используют методы: иммунофенотипирование, определение цитогенетических маркеров.

Иммунофенотипирование ОЛ. На поверхности и в цитоплазме гемопоэтических клеток определены специфические белки – антигены (150), сгруппированные в кластеры дифференцировки (CD). Определение их методом моноклональных антител позволяет определить их линейную принадлежность и этап дифференцировки (при том, что специфических антигенов для лейкозных клеток не обнаружено). Задачи иммунофенотипирования: Подтверждение диагноза. Установление варианта ОЛ (в дополнение к цитоморфологическому методу). Определение бифенотипических вариантов ОЛ. Характеристика аберрантного иммунофенотипа. Выделение прогностических групп.

Цитогенетическая характеристика острых лейкозов. Практически у 90% больных ОЛ находятся генетические поломки, характеризующиеся различными дефектами хромосом. На основании результатов цитогенетических исследований ОЛ в настоящее время классифицированы, выделены их отдельные формы. Определенные цитогенетические маркеры являются принципиально важными в плане как терапии, так и прогноза течения ОЛ. Так, обнаружение филадельфийской (Ph’) хромосомы требует включения в программу терапии препарата гливек (иматиниб), избирательно подавляющего синтез патологического белка транслоцированным участком 21 -й пары хромосом, а, следовательно, и стимулируемое этим белком бесконтрольное размножение клеток. Наличие химерного гена PML/RARα, образующегося в результате транслокации t(15; 17), подтверждает диагноз острого промиелоцитарного лейкоза, в лечении которого обязательно использование препарата трансретиноевой кислоты (ATRA).
. Начальная стадия – стадия масок (случайное выявление при профилактическом осмотре или")
Клиническая классификация (1). Начальная стадия – стадия масок (случайное выявление при профилактическом осмотре или обследовании по поводу какой-либо другой патологии или дебютирование картиной респираторного заболевания, ангины, немотивированной лихорадки, пневмонии, менингита, упорного радикулита, оссалгии или артралгии, увеличения лимфоузлов, гепатоспленомегалии и др. ). При любом неясном или затянувшемся заболевании необходимо производить полный анализ крови!!!
. 2. Развернутая – стадия первой атаки. Выраженное угнетение нормального кроветворения и")
Клиническая классификация (2). 2. Развернутая – стадия первой атаки. Выраженное угнетение нормального кроветворения и высокий бластоз костного мозга.
. 3. Стадия ремиссии. а) полная – в пунктате костного мозга –")
Клиническая классификация (3). 3. Стадия ремиссии. а) полная – в пунктате костного мозга – не более 5% бластов или общее количество бластов и лимфоидных клеток не более 40% (из них бластов – менее 5 %), лейкоцитов в крови – не менее 1, 5 х109/л, тромбоцитов – не менее 100 х109/л, внекостномозговые пролифераты отсутствуют. б) неполная – гематологическое улучшение (существенное снижение % бластных клеток в костном мозге и улучшение состава периферической крови) или исчезновение бластов из крови при сохранении в костном мозге, ликвидация клинических симптомов. в) выздоровление – полная ремиссия на протяжении 5 и более лет.
. 4. Стадия рецидива – 2, 3 атаки. Рецидив может быть костномозговым")
Клиническая классификация (4). 4. Стадия рецидива – 2, 3 атаки. Рецидив может быть костномозговым – более 5% бластов в костном мозге или местным (появление экстрамедуллярного очага кроветворения). В последние годы выделяют цитогенетический вариант рецидива острого лейкоза, при котором хромосомные аномалии в отдельных клонах клеток, исчезнувшие в процессе терапии, обнаруживаются вновь. Наличие цитогенетического рецидива острого лейкоза (даже при констатации полной ремиссии при использовании прочих диагностических методов) требует интенсификации терапии. а) ранний (в течение 6 месяцев) – первичная резистентность, б) поздний – вторичная резистентность. Рецидив, развернувшийся на фоне цитостатической терапии, отличается от развернутого периода (первой атаки): он прогностически более тяжел и требует новой комбинации цитостатических препаратов.
. 5. Терминальная стадия. Ей нередко предшествует частичная ремиссия (лишь!): патологические клетки")
Клиническая классификация (5). 5. Терминальная стадия. Ей нередко предшествует частичная ремиссия (лишь!): патологические клетки становятся менее чувствительны к цитостатикам, чем нормальные клетки – т. е. под влиянием цитостатиков гранулоцитопения нарастает быстрее, чем уменьшается содержание бластных клеток. Это делает невозможным получение полной гематологической ремиссии. В дальнейшем наступает момент, когда все цитостатики не только оказываются неэффективными, но процесс прогрессирует и на их фоне: нарастают цитопении, появляются некрозы слизистых, спонтанные кровоизлияния. Основные признаки терминальной стадии – а) резистентность заболевания к проводимой терапии, б) миелофиброз с панцитопенией в периферической крови, в) возникновение лейкемических очагов в коже, печени, ЦНС, миокарде, почках и других органах.
. Гиперпластический (пролиферативный): увеличение")
В развернутую стадию в клинической картине можно выделить несколько синдромов. 1). Гиперпластический (пролиферативный): увеличение л/у, селезенки, печени, миндалин, боли в костях (гиперплазия костного мозга, лейкозная реакция – отслоение кости от надкостницы). Если увеличенные лимфоузлы средостения сдавливают верхнюю полую вену – отеки шеи, лица, верхних конечностей (синдром верхней полой вены). Гиперплазия десен. Тяжелые язвеннонекротические изменения слизистой рта, глотки, миндалин. На коже – лейкемиды (бляшки).
. Интоксикационный: выраженная общая слабость, утомляемость, снижение массы тела, потливость, лихорадка. Причина – распад")
2). Интоксикационный: выраженная общая слабость, утомляемость, снижение массы тела, потливость, лихорадка. Причина – распад опухолевых клеток.
. Анемический: слабость, бледность, одышка, сердцебиение (100%).")
3). Анемический: слабость, бледность, одышка, сердцебиение (100%).
. Геморрагический: подкожные ( «шкура леопарда» ), подслизистые кровоизлияния, десневые, носовые, маточные и другие")
4). Геморрагический: подкожные ( «шкура леопарда» ), подслизистые кровоизлияния, десневые, носовые, маточные и другие кровотечения (50 -60%). Причина: тромбоцитопения, повышенная сосудистая проницаемость, дефицит факторов свертывания крови (протромбина), усиление фибринолиза. 1020% больных погибают от кровотечения.
. Иммунодефицитный (синдром инфекционных осложнений). Нарушение клеточного и гуморального иммунитета.")
5). Иммунодефицитный (синдром инфекционных осложнений). Нарушение клеточного и гуморального иммунитета.
. 1. Синдром нейролейкемии (15 -20%). Ухудшает прогноз, т. к. цитостатики не")
Внекостномозговые проявления (1). 1. Синдром нейролейкемии (15 -20%). Ухудшает прогноз, т. к. цитостатики не проникают через гематоэнцефалический барьер. При проникновении бластов через гематоэнцефалический барьер – 1 фаза – менингеальные симптомы (ригидность затылочных мышц, судороги, повышение температуры), 2 фаза – энцефалит (головная боль, бред, затоможенность, парез языка, мимических мышц), 3 фаза – менингоэнцефалит, 4 фаза – диэнцефалит (поражение ядер клеток) – сонливость, жажда, полиурия, булимия, повышение АД. Полирадикулоневритическая форма поражения нервной системы – нижние парезы, нарушения черепно-мозговых нервов, чувствительности, исчезновение сухожильных рефлексов.
. 2. Мочеполовая система: цистит, дизурия, система гематурия, простатит, дисменорея. В почках")
Внекостномозговые проявления (2). 2. Мочеполовая система: цистит, дизурия, система гематурия, простатит, дисменорея. В почках могут быть отдельные очаги опухолевого роста и диффузная инфильтрация. Почки увеличены, процесс нередко двусторонний. Возможно развитие почечной недостаточности, анурии. Поражение яичек – преимущественно при лимфобластных формах. Внезапно становятся плотным и увеличивается одно яичко. Поражение возможно и на фоне клинико-гематологической ремиссии. При отсутствии специфической терапии неизбежно и поражение второго яичка.
. 3. ЖКТ: дисфагия, сужение пищевода, язвы ЖКТ желудка, некротический энтероколит. 4.")
Внекостномозговые проявления (3). 3. ЖКТ: дисфагия, сужение пищевода, язвы ЖКТ желудка, некротический энтероколит. 4. Поражение печени: печень увеличена, печени плотная, обычно безболезненная. Причина: инфильтрация бластами, токсическое действие цитостатиков, развитие вирусного гепатита (чаще – вследствие гемотрансфузий), механическая желтуха. 5. Поражение селезенки: увеличение селезенки (инфильтрация бластами, инфаркты ее вен, повышенная нагрузка на селезенку при гиперлейкоцитозе).
. 6. Легкие: лейкозный пневмонит. Речь идет об инфильтрации Легкие бластными клетками")
Внекостномозговые проявления (4). 6. Легкие: лейкозный пневмонит. Речь идет об инфильтрации Легкие бластными клетками межальвеолярных перегородок или образований перибронхиальных муфт, а также о возможном развитии лейкостаза в случаях высокого лейкоцитоза. Клиника: сухой кашель, одышка, повышение температуры, бронхиальное дыхание, сухие хрипы, крепитация над очагами поражения. На рентгенограммах: локальное усиление легочного рисунка, иногда – мелко- или крупноочаговые тени. Выпот в плевральную полость. Диагностика лейкозного пневмонита осложняется тем, что в терминальной стадии при глубокой гранулоцитопении атипично протекает обычная бактериальная пневмония (отсутствие клеточной инфильтрации в очаге воспаления): очень скудные физикальные и рентгенологические данные. Диагностика проводится ex juvantibus: при появлении легочных симптомов назначают антибиотики широкого спектра действия – неэффективность этой терапии доказывает бластную инфильтрацию легких и необходимость смены цитостатика. Лейкозный пневмонит может сочетаться с бактериальной пневмонией.
. 7. Сердечно-сосудистая система: тахикардия, система аритмии, одышка, развитие сердечной недостаточности при")
Внекостномозговые проявления (5). 7. Сердечно-сосудистая система: тахикардия, система аритмии, одышка, развитие сердечной недостаточности при наличии глухости тонов, изменений на ЭКГ. Морфологическая основа – инфильтрация миокарда и скопление в нем бластных клеток.
. 8. Лейкемиды кожи: чаще при миелобластном кожи лейкозе на поздних этапах.")
Внекостномозговые проявления (6). 8. Лейкемиды кожи: чаще при миелобластном кожи лейкозе на поздних этапах. Множественные плотные или мягкие, поднимаются над поверхностью кожи, розового или светлокоричневого цвета, иногда без окраски. Иногда с захватом подкожно-жировой клетчатки и образованием плотных и спаянных с кожей узлов. Гистологически – бластные клетки.
. 9. Кости: боли, переломы. Кости 10. Глаза: светобоязнь, слезотечение, снижение Глаза")
Внекостномозговые проявления (7). 9. Кости: боли, переломы. Кости 10. Глаза: светобоязнь, слезотечение, снижение Глаза зрения, боли. 11. Лейкемическая инфильтрация десен: чаще десен при остром монобластном лейкозе. Десны гиперемированы, нависают над зубами, имеют напоминающие кровоизлияния красные участки. В области лейкемических инфильтратов возможен распад.
. 1. ОАК: анемия нормо- или гиперхромная (а также ОАК анизоцитоз, пойкилоцитоз),")
План обследования (1). 1. ОАК: анемия нормо- или гиперхромная (а также ОАК анизоцитоз, пойкилоцитоз), тромбоцитопения. Увеличение СОЭ. Количество лейкоцитов до 10 х109 – лейкопенический вариант, 10 -50 х109 – сублейкемический вариант, более 50 х109 – лейкемический вариант. Бласты от 3 -5% до 80 -90% и полного замещения. Феномен провала (лейкемическое зияние) – hiatus leukemikus – отсутствие промежуточных форм между бластами и зрелыми лейкоцитами.

2. Стернальная пункция. Настораживает количество бластов более 5%. Пунктат мономорфный, богат клеточными элементами (60 -90%). Если ниже 60% – мультизональные исследования. Менее 30% бластов – малопроцентный лейкоз – вариант миелодиспластического синдрома. При сомнениях – трепанобиопсия (лучше – билатеральная).
. Цитохимические реакции - иммунологическое и тестирование лейкоцитов (иммуноцитохимические и иммуногистохимические исследования).")
План обследования (3). Цитохимические реакции - иммунологическое и тестирование лейкоцитов (иммуноцитохимические и иммуногистохимические исследования). Цитогенетическое исследование клеток костного мозга с целью выявления хромосомных нарушений. Люмбальная пункция с цитологическим исследованием ликвора. УЗИ парензиматозных органов. Рентгенография органов грудной клетки. Биохимия (ЛДГ, фибриноген – повышение, Ал. АТ, Ас. АТ и др. ). Кал на скрытую кровь. Общий анализ мочи.

Дифференциальный диагноз. С хроническими лейкозами, злокачественными лимфомами в стадии лейкемизации, апластической анемией, метастазами рака в костный мозг, лейкемоидными реакциями, инфекционным мононуклеозом.

Лечение. Лечение начинается немедленно после установления диагноза в специализированном стационаре врачом-гематологом. Желательно – асептическая палата. Симптоматическая терапия может быть начата в любом отделении, где был установлен диагноз: антибиотики, гемостатические, дезинтоксикационные средства, трансфузии эритроцитарной массы. 4. Химиотерапия. Основной принцип терапии – быстрое освобождение костного мозга от опухолевых клеток с помощью комбинации цитостатических препаратов в достаточных дозах и за определенный отрезок времени. Максимально полное удаление (эрадикация) лейкозных клеток достигается циклическим применением программ лечения, включающим препараты разной направленности действия. а) индукция (достижение ремиссии). б) консолидация (закрепление) ремиссии. в) профилактика нейролейкемии. г) поддерживающая терапия.

Профилактику нейролейкемии проводят только: при острых лимфобластных лейкозах, при остром миеломонобластном и монобластном, при всех формах острого миелобластного лейкоза с гиперлейкоцитозом. Профилактику проводят при нормальном составе спинномозговой жидкости, полученной при первой пункции. Она включает интралюмбальное введение трех препаратов: метотрексата, преднизолона и цитарабина или краниальное облучение в суммарной дозе 2, 4 Гр. Лечение нейролейкемии проводят при исходно патологическом составе спинномозговой жидкости и используют более жесткие схемы.
 (1). Профилактика инфекционных осложнений: соблюдение санэпидрежима (проведение химиотерапии в условиях")
Симптоматическая терапия (терапия поддерживания) (1). Профилактика инфекционных осложнений: соблюдение санэпидрежима (проведение химиотерапии в условиях изолированной палаты, лучше – боксированной, соблюдение правил асептики и антисептики, стерилизация воздуха), профилактическое применение в период цитопенического синдрома бисетола и стерилизации кишечника неадсорбируемыми антибиотиками, активная антибактериальная и противогрибковая терапия в период глубокого агранулоцитоза и инфекционных осложнений, обработка полости рта и др. Использование ростовых факторов для уменьшения длительности агранулоцитоза. Молграстим (лейкомакс), филграстим, нейпоген (рекомбинантный ГМ-КСФ), граноцид (рекомбинантный ГКСФ) повышают содержание нейтрофилов, 5 мкг/кг массы тела п/к или в/в 2 -3 раза в неделю.
 (2). Профилактика язвенно-некротического энтероколита – полное парентеральное питание в период")
Симптоматическая терапия (терапия поддерживания) (2). Профилактика язвенно-некротического энтероколита – полное парентеральное питание в период агранулоцитоза. Профилактика и лечение анемии (трансфузия анемии эритроцитарной массы: при анемии 3 степени или появлении признаков циркуляторно-гипоксического синдрома при более высоких цифрах гемоглобина – одышки, тахикардии, головокружения, обмороков и др. ). Профилактика и лечение геморрагических осложнений (заместительные трансфузии тромбоцитов (по 4 -6 доз 2 -3 раза в неделю при снижении уровня тромбоцитов ниже 20*109/л даже при отсутствии геморрагий или при снижении уровня тромбоцитов ниже 30*109/л при наличии геморрагического синдрома).
 (3). Предотвращение синдрома «бластного лизиса» – выделение значительного количества пуриновых")
Симптоматическая терапия (терапия поддерживания) (3). Предотвращение синдрома «бластного лизиса» – выделение значительного количества пуриновых соединений при распаде бластов и лейкоцитов, резкое повышение содержания в крови мочевой кислоты с отложением ее кристаллов в почках и мочевых путях. Назначают препараты, выводящие соли мочевой кислоты (милурит) или тормозящие ее синтез (аллопуринол). Рекомендуется приступать к лечению аллопуринолом при повышении содержания мочевой кислоты в крови более 0, 3 г/л. В целях предупреждения выпадения кристаллов мочевой кислоты в мочевыводящих путях следует употреблять большие количества щелочных жидкостей.

Прочее лечение Применение интерферона, обладающего антипролиферативным и антивирусным эффектами, усиливающего активность Т-лимфоцитов-киллеров по отношению к лейкозным (бластным) клеткам. Профилактика тошноты и рвоты (эметрон, эмесет в/в перед введением цитостатиков). Профилактика изъязвления желудка и 12 -перстной кишки вследствие применения глюкокортикоидов – применение ингибиторов протоновой помпы (ультоп, омепразол), Н 2 -блокаторов (квамател, фамотидин), антацидов (маалокс, альмагель). Профилактика электролитных нарушений, особенно на фоне калийвыводящих препаратов (мочегонные, амфотерцин В и др. ).
")
Трансплантация костного мозга – аллогенная (от родного брата или сестры, подобранного по системе HLA) и аутогенная (собственный костный мозг, заготовленный в период ремиссии). Эффективность аллогенной трансплантации выше, позволяет добиться 5 летней выживаемость у 60% перенесших ее пациентов, но возможны тяжелые осложнения (отторжение трансплантанта, тяжелые инфекционные осложнения, веноокклюзионная болезнь) – смертность 20 -25%. Аутологичная трансплантация переносится легче, смертность 10%, 50% рецидивов. Преимущество аутологичной трансплантации перед химиотерапией – возможность прекращения лечения. Показания к аллогенной трансплантации – неблагоприятные факторы прогноза, в фазе первой ремиссии, возраст до 40 лет, хороший соматический статус.

Спорные моменты терапии ОЛ До сих пор отсутствует единое мнение о значимости курсов консолидации ремиссии, длительной химиотерапии поддержания ремиссии, этапов ранней и поздней интенсификации лечения в постремиссионной терапии острых лейкозов. Показано, что соблюдение принципа химиотерапии «доза-эффект» на этапе постремиссионного лечения имеет принципиальное значение для показателей выживаемости больных острым лейкозом, т. е. попытки уменьшения длительности применения химиопрепаратов или снижения их доз приводят к возрастанию частоты рецидивов заболевания, и, следовательно, смертности больных. Поддерживающая цитостатическая терапия прерывается при содержании лейкоцитов менее 1 х109/л и возобновляется при повышении их количества более 2, 5 х109/л. Длительность химиотерапии в ремиссии очень различна: от 3 месяцев до 7 лет по данным различных авторов. С учетом разных данных о сроках наступления цитогенетической ремиссии желательно проведение активного лечения не менее 6 месяцев с момента установления полной клинико-гематологической ремиссии, а при невозможности контролировать цитогенетический ответ – не менее 12 месяцев. Использование трансплантации костного мозга после достижения первой ремиссии позволяет не только улучшить прогноз, но и уменьшить длительность терапии (до 7 -10 месяцев от момента начала химиотерапии в период первой атаки).

ХРОНИЧЕСКИЕ ЛЕЙКОЗЫ Самыми частыми вариантами хронических лейкозов являются: Хронический миелолейкоз. Хронический лимфолейкоз. Эссенциальная эритремия (истинная полицитемия). Множественная миелома.

ХРОНИЧЕСКИЙ МИЕЛОЛЕЙКОЗ Опухоль системы крови, возникающая из клетки- предшественницы миелопоэза, которая сохраняет способность дифференцироваться до зрелых форм. Субстрат – молодые и созревающие нейтрофилы. Хронический миелолейкоз (ХМЛ), называемый также хроническим миелобластным или хроническим миелоцитарным лейкозом, самая часто встречаемая разновидность лейкоза (20% от всех лейкозов). Ежегодная заболеваемость составляет 1 -1, 5 случая на 100 000 населения. В России каждый год фиксируется около 2500 новых случаев этого заболевания. ХМЛ чаще болеют взрослые, около 2% случаев приходится на долю детей. Чаще болеют мужчины 30 -70 лет. В 86 -90% случаев – появление в клетках филадельфийской (Ph) хромосомы – 22 аутосомы с укороченным длинным плечом вследствие реципрокной транслокации между 9 и 22 хромосомами.

Филадельфийская хромосома

Классификация ХМЛ. Начальная стадия – миелоидная пролиферация костного мозга с изменениями в периферической крови без явлений интоксикации. Начальная стадия практически не диагностируется. Это случаи, когда лишь небольшая часть клеток костного мозга оказывается с Ph’-хромосомой, а основная – без этой хромосомы. В основном болезнь определяется в хронической стадии.

1. Хроническая стадия – выраженные клинико-гематологические проявления – постепенно нарастающий лейкоцитоз – от 15 -20 х109/л до 500 -900 х109/л, увеличение селезенки, которая без лечения часто достигает громадных размеров и в которой часто развиваются инфаркты. Больные постепенно теряют массу тела, нарастают дистрофические изменения органов, появляется анемический синдром. Даже пациенты молодого возраста в процессе развития заболевания (чаще через 3 -4 года) полностью утрачивают трудоспособность и нуждаются в постороннем уходе. Доброкачественная моноклоновая опухоль превращается в поликлоновую – процесс вступает в стадию акселерации.

2. Стадия акселерации характеризуется прежде всего снижением чувствительности к ранее эффективной терапии. В крови нарастает количество незрелых элементов гемопоэза (промиелоцитов, метамиелоцитов), суммарное число которых начинает превышать количество зрелых элементов, чего никогда не бывает в хронической стадии болезни. Усиливается общая симптоматика. Международными признаками стадии акселерации являются добавочные хромосомные аберрации (помимо Ph-хромосомы), обнаружение 15% или более бластов в крови или 30% и более в сумме бластов и промиелоцитов в костном мозге, 20% базофилов в крови, падение уровня тромбоцитов ниже 100 х109/л. Эта стадия может продолжаться от 2 -3 месяцев до 1, 0 -1, 5 лет.

3. Терминальная стадия – рефрактерность к проводимой цитостатической терапии; в 80 -85% случаев – развитие бластного криза (по лимфоидному или миелоидному варианту) или экстрамедуллярных саркоматозных очагов; значительное увеличение селезенки и (у многих) печени; появление жалоб на боли в костях, мышцах, суставах; резкие подъемы температуры с ознобами и проливными ночными потами; снижение массы тела; выраженная общая слабость. Эта стадия до недавнего времени даже при лечении продолжалась не более полугода.
, меньше – печени); интоксикация.")
Клинические синдромы: 1. Миелопролиферативный (боли в костях, увеличение селезенки (особенно), меньше – печени); интоксикация. Некрозов и лейкемидов нет! 2. Анемический синдром. 3. Тромбо-геморрагический синдром (слабо выражен). 4. Уратовые диатезы (распад нейтрофилов).
. Красная кровь в начале болезни существенно не меняется, затем по мере")
Картина крови (1). Красная кровь в начале болезни существенно не меняется, затем по мере вытеснения нормальных ростков кроветворения и миелотоксического действия специфического лечения развивается анемия. Тромбоциты вначале повышаются (у 1/3 больных – до высоких цифр – 1500 -2000*109/л), затем снижаются.
. В хронической стадии картина крови характеризуется нейтрофильным лейкоцитозом со сдвигом влево.")
Картина крови (2). В хронической стадии картина крови характеризуется нейтрофильным лейкоцитозом со сдвигом влево. Количество лейкоцитов 12 -15*109/л, реже – более 20*109/л. При лейкоцитозе, превышающем 20 -30*109/л возникают слабость, потливость, повышенная утомляемость. Сдвиг влево происходит до миелоцитов и промиелоцитов (редко – бластов), которые представлены единицами (промиелоциты и миелобласты до 5%, юные повышены). Палочек более 6%, сегментов менее 45%, моноциты и лимфоциты снижены. Базофильно-эозинофильная ассоциация – увеличение абсолютного числа базофилов и эозинофилов в крови, чаще – при одномоментном повышении их процентного содержания (базофилов более 1%, эозинофилов более 5%). Без лечения лейкоцитоз неуклонно растет, количество тромбоцитов или стабильно, или неуклонно увеличивается.
. По мере прогрессирования процесса (переход через стадию акселерации в терминальную")
Картина крови (3). По мере прогрессирования процесса (переход через стадию акселерации в терминальную стадию) в крови нарастает анемия, тромбоцитоз (реже) или тромбоцитопения, количество лейкоцитов может снижаться (при экстрамедуллярной локализации химиорезистентных лейкоцитов) или возрастать (при костномозговой их локализации), нарастает сдвиг нейтрофильной формулы влево вплоть до высокого бластоза и лейкемического провала, сохраняется и нарастает базофильноэозинофильная ассоциация.

18, 0 5 8 2 4 9 22 мм/ч 32 20 10

64, 2 1 3 1 5 9 43 мм/ч 45 21 5

22, 5 0 3 1 3 5 19 мм/ч 62 22 5

12, 0 1 1 2 2 2 Бласты 70% 23 мм/ч 15 5 2

12, 0 2 2 4 4 6 Бласты 5% 25 мм/ч 42 26 9

0, 93 2, 5*10/л 85 г/л 0, 1% 24*10/л

1, 0 4, 2*10/л 140 г/л 0, 1% 425*10/л
.")
В костном мозге – миелоидная пролиферация (соотношение лейко/эритро 10: 1 – 20: 1). В трепанате – практически полное вытеснение жирового костного мозга кроветворными клетками, преимущественно гранулоцитами. При высоком тромбоцитозе периферической крови в трепанате выявляется большое количество мегакариоцитов.

Активность щелочной фосфатазы нейтрофилов снижена. Реакция на миелопероксидазу в миелоцитах, промиелоцитах, зрелых нейтрофилах сохранена, хотя и снижена.

Лечение хронического миелолейкоза началось с середины XIX века с применения мышьяка, который несколько уменьшал размеры селезенки и улучшал самочувствие больных, но не влиял на продолжительность жизни. С начала ХХ века в лечении стало широко применяться облучение селезенки, а затем и всего тела, которые приводили в регрессии симптоматики на короткий срок, причем повторные курсы лучевой терапии были все менее эффективными.

В настоящее время применяют следующие методы лечения больных хроническим миелолейкозом: трансплантация костного мозга, химиотерапия, альфа – интерферон, гливек (иматиниб), симптоматическая терапия.


Хронический моноцитарный лейкоз относится к редким формам лейкозов, характеризуется высоким моноцитозом в периферической крови (20 -40%) при нормальном или несколько повышенном числе лейкоцитов. Наряду со зрелыми моноцитами в крови имеются единичные промоноциты. В костном мозге процент моноцитов повышен незначительно, но в трепанате наблюдается гиперплазия костномозговой ткани с диффузным разрастанием моноцитарных элементов. В крови и моче высокое содержание лизоцима. У 50% больных пальпируется селезенка. Длительное благополучное течение хронического моноцитарного лейкоза может смениться терминальной стадией, имеющей те же особенности, что и терминальная стадия хронического миелолейкоза. В развернутой стадии процесс не требует специального лечения, только при глубокой анемии необходимо периодическое переливание эритроцитарной массы, которое можно проводить амбулаторно.
. ХЛЛ – В-клеточное лимфопролиферативное заболевание, морфологическим субстратом которого является клон лимфоцитов,")
ХРОНИЧЕСКИЙ ЛИМФОЛЕЙКОЗ (ХЛЛ). ХЛЛ – В-клеточное лимфопролиферативное заболевание, морфологическим субстратом которого является клон лимфоцитов, имеющих размеры и морфологию нормального лимфоцита и иммунофенотип, соответствующий иммунофенотипу В-лимфоцита поздних стадий дифференцировки. Т-клеточные ХЛ – это Т-пролимфоцитарный лейкоз (не более 2% от всех ХЛЛ) и лейкоз из больших гранулярных лимфоцитов (23% от всех ХЛЛ). Также выделяют В-пролимфоцитарный ХЛЛ (не более 1% от всех ХЛЛ) В-ХЛЛ – наиболее распространенный вид лейкозов в странах Европы и Северной Америки, на его долю приходится около 30% среди всех лейкозов. Ежегодная заболеваемость составляет 3 -3, 5 на 100 000 населения, повышаясь для лиц старше 65 лет до 20 на 100 000.

ХЛЛ традиционно считается заболеванием лиц пожилого возраста. Действительно, в странах Западной Европы и США средний возраст на момент установления диагноза увеличился с 60 -65 до 70 лет, что обусловлено как увеличением общепопуляционной продолжительности жизни в этих странах, так и улучшением диагностики ХЛЛ. Лишь 10% заболевают в возрасте до 40 лет. Мужчины : женщины = 2 : 1. Диагноз ХЛЛ должен быть заподозрен при абсолютном числе лимфоцитов в ОАК 5 х 109/л и более, что обычно соответствует 65 -70% лимфоцитов в лейкоцитарной формуле. Согласно современным международным критериям (Cheson B. B. D. , Bennett J. M. , Grever M. et al, 1996), для установления диагноза ХЛЛ должны присутствовать 3 признака: А. абсолютный лимфоцитоз крови 5 х 109/л и более, В. не менее 30% лимфоцитов в костномозговом пунктате, С. наличие в крови и костном мозге В-клона лимфоцитов с определенным иммунофенотипом. При ХЛЛ патологические лимфоциты экспрессируют на своей поверхности антигены CD 5, CD 19, CD 23, отмечается слабая экспрессия на поверхности клеток иммуноглобулинов (обычно экспрессируются только Ig. M, но изредка – одновременно Ig. D) с одной легкой цепью, определяется слабая экспрессия антгенов CD 20 и CD 22, у ряда больных экспрессируется молекула FМC 7.

Классификация ХЛЛ: Существуют 2 классификации стадийности ХЛЛ – принятая в США классификация K. Rai и распространенная в Европе и наиболее часто используемая в нашей стране классификация J. Binet.
 0 Имеется только лимфоцитоз – более 15 х")
Стадия Характеристика ХЛЛ (по K. Rai) 0 Имеется только лимфоцитоз – более 15 х 109/л в крови и более 40% в костном мозге 1 Имеется указанный лимфоцитоз и увеличение л/у 2 Лимфоцитоз и сплено- и гепатомегалия независимо от увеличения л/у 3 Лимфоцитоз и снижение уровня гемоглобина ниже 110 г/л независимо от увеличения л/у и органов 4 Лимфоцитоз и снижение количества тромбоцитов ниже 100 х 109/л независимо от увеличения л/у и органов
 А При наличии лимфоцитоза крови и костного мозга, позволяющего")
Характеристика ХЛЛ (по J. Binet) А При наличии лимфоцитоза крови и костного мозга, позволяющего установить диагноз, содержание гемоглобина более 100 г/л, тромбоцитов – более 100 х 109/л, имеется увеличение л/у в 1 -2 областях В Содержание гемоглобина и тромбоцитов такое же, как в стадии А, но имеется увеличение л/у в 3 и более областях С Содержание гемоглобина ниже 100 г/л, тромбоцитов – менее 100 х 109/л при любом количестве зон с увеличенными л/у и независимо от увеличения органов

β-2 микроглобулин и ЛДГ Наибольшее прогностическое значение из наиболее доступных в повседневной клинической практике исследований имеют β-2 микроглобулин и ЛДГ показано, что при нормальном уровне ЛДГ в момент постановки диагноза продолжительность жизни составляет 10 -12 лет, при уровне выше нормы в 1, 5 -2 раза – всего 4 -5 лет. Повышение уровня β 2 микроглобулина является признаком высокой пролиферативной активности и быстрого прогрессирования заболевания. При нормальном уровне (3, 0 -3, 5 мг/л) продолжительность жизни колеблется от 8 до 12 лет, в то время как при повышенном в 2 раза – всего 4 -5 лет.

24, 0 1 0 4 0 1 33 мм/ч 24 65 5

72, 0 0 0 1 0 0 24 72 3 Тени Боткина-Гумпрехта 37 мм/ч

8, 6 1 0 0 0 1 26 61 Плазматические клетки 7 40 мм/ч 4

5, 2 1 0 2 0 1 Бласты 23% 23 мм/ч 7 64 2

2, 8 1 0 2 0 1 22 мм/ч 27 61 7

1, 0 4, 2*10/л 140 г/л 0, 1% 425*10/л

0, 9 1, 6*10/л 52 г/л Не обнаруж. 44*10/л
: 1. Доброкачественная – продолжительность жизни больных – 30 -40 лет. 2.")
Клинико-лабораторные варианты (формы): 1. Доброкачественная – продолжительность жизни больных – 30 -40 лет. 2. Прогрессирующая (классическая) форма, при которой лейкоцитоз и размеры лимфоидных органов увеличиваются значительно быстрее, раньше развиваются осложнения. Средняя продолжительность жизни – не более 6 -8 лет. 3. Опухолевая – протекающая с преимущественным увеличение лимфоузлов, 4. Спленомегалическая, 5. Костномозговая, 6. ХЛЛ, осложненный цитолитическим синдромом, 7. ХЛЛ, протекающий с парапротеинемией, 8. Волосатоклеточный лейкоз – характеризуется морфологическими (выросты цитоплазмы, напоминающие ворсинки), цитохимическими (резко положительная реакция на кислую фосфатазу, не подавляющаяся тартратом натрия), клиниколабораторными особенностями (лейкопения, выраженная спленомегалия), высокой эффективностью лечения альфа-интерфероном. 9. Т-форма (встречается практически исключительно в Японии) – типичны более молодой возраст больных, специфическая лимфоидная инфильтрация кожи, нарушения кариотипа, быстро прогрессирующее течение и неблагоприятный прогноз.
, интоксикация. Сыпь, лейкемиды мало характерны.")
Клинические синдромы: 1. Лимфопролиферативный – увеличение гемопоэтических органов (л/у), интоксикация. Сыпь, лейкемиды мало характерны. Картина крови: эритроциты, тромбоциты, эозинофилы, базофилы, палочки, сегменты, моноциты снижены. Лейкоцитоз более 20 х109/л, лимфоцитов более 40%. Тени Боткина-Гумпрехта, они же клетки лейколиза или цитолиза (ядра раздавленных стеклом нестойких лимфоцитов). В начале болезни в крови пролимфоцитов и лимфобластов нет. Однако в некоторых случаях пролимфоциты преобладают в крови с начала – пролимфоцитарная форма ХЛЛ. По мере развития болезни в крови начинают встречаться единичные пролимфоциты и лимфобласты. Большое их количество появляется лишь в терминальной стадии болезни. Миелограмма – лимфоцитоз более 30%.

2. Анемический – в т. ч. по причине аутоиммунной гемолитической анемии. 3. Геморрагический – слабо выражен. 4. Инфекционные осложнения, часто вирусные.
: 1. Инфекционные – нарушения гуморального и клеточного иммунитета (снижение иммуноглобулинов А,")
Осложнения ХЛЛ (1): 1. Инфекционные – нарушения гуморального и клеточного иммунитета (снижение иммуноглобулинов А, М, G, гипогаммаглобулинемия. Связана с нарушением взаимодействия Т и В-лимфоцитов, повышением Т -супрессоров, неспособностью лейкозных Влимфоцитов отвечать на лимфокины, выделяемые Т-клетками). Инфекционные осложнения – основная причина смерти (воспаление легких, флегмоны, герпетическая инфекция). 2. Выраженная неадекватная реакция на укус насекомых – значительная инфильтрация в месте укуса комаров с тяжелой интоксикацией.
: 2. Цитопении (анемия, тромбоцитопения). Генез различен – тотальная лимфоидная инфильтрация костного")
Осложнения ХЛЛ (2): 2. Цитопении (анемия, тромбоцитопения). Генез различен – тотальная лимфоидная инфильтрация костного мозга, гиперспленизм, аутоантитела к эритроцитам и/или тромбоцитам. 3. Злокачественная трансформация. Переход заболевания в неходжкинскую лимфому высокой степени злокачественности (синдром Рихтера), развитие второй злокачественной гематологической (острый лейкоз, миеломная болезнь) или негематологической (рак) опухоли. Это обусловлено нарушениями противоопухолевого иммунитета и проводимой цитостатической терапией. 4. Нейролейкемия. Часто – поражение 8 пары ЧМН с ослаблением слуха, корешковый синдром. 5. Иммунокомплексные осложнения – синдром Шегрена, Шейнлейн-Геноха. Тяжелая почечная недостаточность (внезапная анурия) – инфильтрация паренхимы опухолевыми клетками.

Больные умирают главным образом от инфекционных осложнений, истощения, геморрагий, анемии, саркомного роста.
. Режим труда и отдыха, витаминотерапия.")
Лечение ХЛЛ: длительное наблюдение с контролем гемограммы (выжидательная тактика). Режим труда и отдыха, витаминотерапия. Показания к началу лечения – ухудшение общего состояния, быстрое увеличение л/у, селезенки, печени, нарастающий лейкоцитоз (более 100 х109/л), развитие аутоиммунной анемии или тромбоцитопении, увеличенная подверженность бактериальным инфекциям. Поскольку ХЛЛ является болезнью накопления долгоживущих, неделящихся зрелых лимфоцитов, вплоть до конца ХХ века проводилась терапия, лишь сдерживающая прогрессирование заболевания.
 – хронический лейкоз, возникающий из клеткипредшественницы миелопоэза, сохранившей")
ЭРИТРЕМИЯ. Истинная полицитемия (эритремия, болезнь Вакеза) – хронический лейкоз, возникающий из клеткипредшественницы миелопоэза, сохранившей способность дифференцироваться по нескольким росткам кроветворения (эритроидному, гранулоцитарному, мегалоцитарному), но преимущественно по эритроидному. Заболеваемость 0, 6 -1, 6 на 100 000. Чаще болеют люди 50 -60 лет. Эритремия – наиболее доброкачественное системное заболевание кроветворных органов. В основе заболевания – лейкозная пролиферация трех ростков кроветворения. В связи с тем, что доминирует гиперплазия эритроидной ткани, основной субстрат опухоли составляют созревающие эритроциты. Увеличенное количество циркулирующих эритроцитов и тромбоцитов снижает скорость кровотока, повышает вязкость и свертывающий потенциал крови.
: плеторический и миелопролиферативный синдром. Плеторический синдром: 1. Субъективные проявления: -")
Клиника истинной полицитемии (1): плеторический и миелопролиферативный синдром. Плеторический синдром: 1. Субъективные проявления: - головные боли, - головокружение, - стенокардия, - тяжесть в голове (вышеперечисленные симптомы связаны с усилением кровенаполнения).
: 2. Объективные проявления: - покраснение кожных покровов с синюшным оттенком")
Клиника истинной полицитемии (1): 2. Объективные проявления: - покраснение кожных покровов с синюшным оттенком (эритроцианоз), - инъекция сосудов склер ( «кроличьи глаза» ), - спленомегалия (вследствие повышения кровенаполнения. Спленомегалия вследствие миелоидной метаплазии селезенки является проявлением миелопролиферативного синдрома), - кожный зуд, усиливающийся после водных процедур (при распаде увеличенного количества эритроцитов выделяется большое количество пуринов, мочевой кислоты, которые раздражают нервные окончания, вызывая мучительный кожный зуд), - повышение АД (обусловлено повышением периферического сопротивления в ответ на повышение вязкости крови, хроническим пиелонефритом, нарушением кровообращения (тромбоз, склероз артерий) в почках). Повышение АД резистентно к стандартной антигипертензивной терапии. АД снижают путем кровопускания, - эритромелалгия – острые жгучие боли в кончиках пальцев, купирующиеся приемом аспирина. Эритромелалгия может проявляться своеобразным отеком с ограниченной гиперемией нижней трети голени или стопы, сопровождающимся резкой болезненностью.

Плетора является главной причиной сосудистых осложнений – тромбозов и кровотечений. Тромбозы (в том числе эритромелалагия, ишемия и даже гангрена пальцев, стенокардия и инфаркты органов, нарушения зрения, динамические нарушениями мозгового кровообращения, инсульты и т. д. ) обусловлены нарушениями микоциркуляции за счет повышения вязкости крови, замедления кровотока, увеличения массы эритроцитов, локального внутрисосудистого или диссеминированного внутрисосудистого свертывания. Наряду с тромботическими тенденциями почти постоянно имеется потенциальная опасность кровотечений, которая проявляет себя при любых, даже малых, оперативных вмешательствах. Обычны длительные кровотечения после экстракции зубов. Спонтанные кровотечения редки, но возможны, особенно из сосудов желудочно-кишечного тракта и геморроидальных узлов. Причина кровотечений – относительная гипофибриногенемия, функциональная неполноценность тромбоцитов, нарушение ретракции кровяного сгустка и феномен «ускользания» эритроцитов: крупнопетлистая сеть эритроцитов не в состоянии удерживать в сгустке (красном тромбе) всю массу эритроцитов, и они «уходят» из рыхлого тромба.

Повышенная гибель клеток на ядерных предстадиях лежит в основе эндогенной урикемии и урикозурии, встречающихся у 40 -50% больных. Клиническими проявлениями нарушений обмена мочевой кислоты являются мочекаменная болезнь и подагра. Клиника развивается постепенно. В анамнезе задолго до установления диагноза имеются указания на кровотечения при экстракции зубов, кожный зуд после ванны и «хорошие» показатели красной крови. Затем нарастает покраснение кожи, слабость, тяжесть в голове, повышение АД, увеличение селезенки, эритромелалгия, кожный зуд, усиливающийся после водных процедур.
: I – начальная стадия – 5 лет и более –")
Классификация истинной полицитемии (1): I – начальная стадия – 5 лет и более – умеренная плетора, селезенка не пальпируется, умеренный эритроцитоз в ОАК, в костном мозге – панмиелоз или гиперплазия эритроцитарного ряда. Сосудистые и висцеральные осложнения – редко.
: II A – полицитемическая – 5 -10 -15 лет, выраженный")
Классификация истинной полицитемии (2): II A – полицитемическая – 5 -10 -15 лет, выраженный плеторический синдром, сплено- и гепатомегалия, тромбозы артерий и вен, геморрагические осложнения. Спленомегалия обусловлена плеторой, отсутствует миелоидная метаплазия селезенки. Кровотечения могут привести к дефициту железа. ОАК – нарастающий эритроцитоз, тромбоцитоз и нейтрофилез с палочкоядерным сдвигом. Трепанобиоптат – панмиелоз, мегакариоцитоз, участки очагового миелофиброза.
: II Б – полицитемическая с миелоидной метаплазией селезенки – нарастающая")
Классификация истинной полицитемии (3): II Б – полицитемическая с миелоидной метаплазией селезенки – нарастающая сплено- и гепатомегалия. Доказательство миелоидной метаплазии селезенки – пункция селезенки и гистологическое исследование биоптата. В клинической картине появляются аллергические проявления, уратовый диатез, истощение, тромбозы, кровоточивость. Наибольшую угрозу во II А и Б стадиях представляют тромбозы. Панцитоз периферической крови со сдвигом лейкоцитарной формулы до миелоцитов, панмиелоз костного мозга и более выраженный миелофиброз. По мере прогрессирования процесса в крови могут в значительном количестве появляться незрелые эритроциты и лейкоциты, нарастает анизо- и пойкилоцитоз: появляются микроциты, овалоциты и эритроциты каплевидной формы.
: III – анемическая – исход заболевания – нарастающая анемия, тромбоцитопения")
Классификация истинной полицитемии (4): III – анемическая – исход заболевания – нарастающая анемия, тромбоцитопения или панцитопения, выраженная сплено- и гепатомегалия, распространенный миелофиброз костного мозга с сохраненным или редуцированным миелопоэзом. От начала болезни проходит 15 -20 лет. Развитию анемической стадии предшествует увеличение селезенки, уменьшение плеторы, появление лейкоэритробластоза периферической крови. Исходом полицитемии может быть острый лейкоз (чаще миелобластный или эритробластный), хронический миелолейкоз, апластическая анемия.

Критерии диагноза истинная полицитемия. Критерии диагноза. А: - увеличение массы циркулирующих эритроцитов; - нормальное насыщение крови кислородом; - спленомегалия. В: - лейкоцитоз более 12 тысяч (без инфекций, интоксикаций); - тромбоцитоз более 400 тысяч (при отсутствии кровотечения); - повышение содержания щелочной фосфатазы нейтрофилов (без инфекций); - повышение витамин В 12 -связывающей способности сыворотки. Диагноз эритремии достоверен при 2 положительных признаках А и В или всех 3 -х критериях А. При плеторе, спленомегалии, лейко- и тромбоцитозе диагноз ясен, но все равно обязательна трепанобиопсия подвздошной кости для подтверждения диагноза.

1, 0 6, 9*10/л 230 г/л 0, 1% 625*10/л

18, 0 1 3 0 0 9 1 мм/ч 64 17 6

0, 95 6, 8*10/л 224 г/л 0, 1% 175*10/л

1, 0 2, 2*10/л 72 г/л 0, 1% 125*10/л

Диагностические трудности Трудности обычно возникают при чисто эритроцитемических формах эритремии – без спленомегалии, лейко- и тромбоцитоза. Для целей дифференциальной диагностики используется определение массы циркулирующих эритроцитов в сочетании с объемом плазмы, определение содержания эритропоэтинов, исключаются заболевания (почки!), часто сопровождающиеся эритроцитозом.

Дифференциальный диагноз истинной полицитемии – Дифференциальный диагноз – с абсолютными и относительными симптоматическими (т. е. вторичными) эритроцитозами. Абсолютный эритроцитоз характеризуется повышенным эритропоэзом и увеличением объема циркулирующих эритроцитов. Встречается: 1 а. При тканевой генерализованной гипоксии: с артериальной гипоксемией (у жителей высокогорья, при высотной болезни, заболеваниях легких, врожденных пороках сердца синего типа), курении (карбоксигемоглобинемия); без артериальной гипоксемии (гемоглобинопатии с повышенным сродством к кислороду). 1 б. При некоторых опухолях, продуцирующих эритропоэтин-подобные субстанции (гипернефрома, опухоли надпочечников, гепатома, гемангиобластома мозжечка, лейомиома матки), 1 в. При локальной ишемии почек (поликистоз, гидронефроз, стеноз почечных артерий). В крови при абсолютных эритроцитозах определяется повышение уровня эритроцитов, гемоглобина, замедление СОЭ (дифференцировать с начальной стадией эритремии).
 – уменьшение объема плазмы и относительное преобладание клеточного состава в единице")
Относительные эритроцитозы (гемоконцентрационные) – уменьшение объема плазмы и относительное преобладание клеточного состава в единице объема крови – повышенная потеря жидкости: 1. с рвотой (инфекции, интоксикации, лучевая болезнь, гестозы, язвенная болезнь желудка и 12 -перстной кишки, передозировка рвотных средств), 2. с диареей (инфекции, интоксикации, передозировка слабительных), 3. через кожу (ожоговая болезнь, гипергидроз при тяжелых инфекциях, при интенсивной нагрузке – марафон, работа в горячем цеху), 4. с мочой (несахарное мочеизнурение, передозировка мочегонных). Кроме того, к относительному эритроцитозу может привести артериальная гипертония, неврастения, стресс-эритроцитоз (синдром Гайсбёка). В крови при относительных эритроцитозах определяется панцитоз и замедление СОЭ (дифференцировать с развернутой стадией эритремии).

Лечение истинной полицитемии. Без лечения 50% больных с клинически выраженной полицитемией умирает в течение 18 месяцев после установления диагноза. Терапия позволяет увеличить продолжительность выживания в среднем на 7 -15 лет. Наиболее частая причина смерти – тромбозы, реже – кровотечения и развитие острого лейкоза.
 и химиотерапия. Кровопускания – при наличии плеторического синдрома")
Патогенетическая терапия истинной полицитемии– кровопускания (гемоэксфузии) и химиотерапия. Кровопускания – при наличии плеторического синдрома и изолированного эритроцитоза. Кровопускания выполняют с целью нормализации количества гемоглобина и показателя гематокрита. Уменьшают зуд, уратовые диатезы и др. , мало влияют на размеры селезенки. Преимущество – отсутствие лейкозогенного действия. Показания: v недавняя и доброкачественно текущая эритремия, v чисто эритроцитемический вариант, v детородный возраст, v рецидивы после цитостатической терапии со снижением лейкоцитов и тромбоцитов. Противопоказания – тромбозы в острой стадии.

Методика кровопусканий: у молодых – 300 -450 мл крови через день, у пожилых – 300 -350 мл через 2 -3 дня. До – 500 мл реополиглюкина с 5000 ЕД гепарина, антикоагулянты. Число процедур определяют показатели крови. Не менее чем за 2 недели до кровопускания назначают курантил 75 -225 мг/сутки, аспирин 100 -125 мг/сутки на постоянный прием или хотя бы 1 -2 недели после кровопускания. Возможны реактивный тромбоцитоз, тромбозы, дефицит железа. При развитии железодефицита после кровопусканий препараты железа лучше не назначать, т. к. они ускоряют прогрессирование заболевания. При выраженном железодефиците и плохой его переносимости назначать препараты железа с цитостатиками. Эритроцитаферез – интервалы 5 -7 дней.

Цитостатическая терапия. Показания: II Б стадия, панцитоз, сосудистые и висцеральные осложнения, тяжелое состояние больного, неэффективность и плохая переносимость кровопусканий. Еженедельный гематологический контроль. Не допускать снижение лейкоцитов ниже 5 х109/л и тромбоцитов ниже 100 х109/л. Алкилирующие (имифос, миелосан, миелобромол, цитостоп, алкеран) и антиметаболиты (гидроксимочевина – преимущественно, 6 меркаптопурин – при бластемии). Цитостатики назначают до нормализации красной крови, затем отменяют. Поддерживающая терапия цитостатиками не рекомендуется из-за малой эффективности и лейкозогенного действия.

Симптоматическая терапия – гипотензивные (мочегонные противопоказаны, желательно – ингибиторы АПФ с кардио-, нефро-, ангиопротекторной целью), антигистаминные (при зуде), аллопуринол (при уратовом диатезе), дезагреганты, антикоагулянты. При аутоиммунной гемолитической анемии или тромбоцитопении – глюкокортикоиды. Выраженная спленомегалия с гиперспленизмом – лучевая терапия на селезенку. При развитии миелофиброза с панцитопенией (III стадия) – заместительная гемокомпонентная терапия. При исходе в острый лейкоз или хронический миелолейкоз – соответствующая химиотерапия (эффективность низкая).

МНОЖЕСТВЕННАЯ МИЕЛОМА. Синонимы: миеломная болезнь, болезнь Рустицкого Калера. Самый частый парапротеинемический гемобластоз. Множественная миелома – злокачественная пролиферация плазматических клеток (т. е. Влимфоцитов, которые дифференцируются до стадии, когда они способны к выработке иммуноглобулинов) главным образом в костном мозге, а также в других системах органов. Клетки могут образовывать солитарные опухоли, известные как плазмоцитомы. Продуцируют патологический иммуноглобулин в больших количествах, откладывающийся в разных органах (печень, почки, сердце) и способствующий повышению вязкости крови. Выход плазматических клеток в периферическую кровь нехарактерен для этого лейкоза, хотя иногда возможен.
: Существует несколько взаимодополняющих классификаций множественной миеломы. I. Клинико-анатомическая классификация: :")
Классификация множественной миеломы (1): Существует несколько взаимодополняющих классификаций множественной миеломы. I. Клинико-анатомическая классификация: : диффузно-очаговая форма (60%); диффузная форма (24%); множественно-очаговая форма (15%); редкие формы (склерозирующая, висцеральная – 1%).
: II. Международная классификация (B. Durie, S. Salmon, 1975): в соответствии")
Классификация множественной миеломы (2): II. Международная классификация (B. Durie, S. Salmon, 1975): в соответствии с которой заболевание делится на три стадии в зависимости от величины опухолевой массы, определяемой по совокупности уровня гемоглобина, кальция, рентгенологических показателей и уровня М-компонента. Кроме того, каждая стадия разделяется на А и В в зависимости от уровня креатинина сыворотки крови. I стадия. Совокупность признаков: уровень гемоглобина более 100 г/л, нормальный уровень кальция сыворотки, отсутствие остеолиза или солитарный костный очаг, низкий уровень М-компонента: Ig. G менее 50 г/л, Ig. A менее 30 г/л, белок Бенс-Джонса в моче менее 4 г/сутки. Опухолевая масса менее 0, 6 кг/м 2 – низкая (1012 клеток составляют 1 кг опухоли). II стадия. Показатели средние между I и III стадиями. Опухолевая масса 0, 6 -1, 2 кг/м 2.
: III стадия. Совокупность признаков: уровень гемоглобина менее 85 г/л, уровень")
Классификация множественной миеломы (3): III стадия. Совокупность признаков: уровень гемоглобина менее 85 г/л, уровень кальция сыворотки выше нормы, выраженный остеодеструктивный процесс, высокий уровень М-компонента: Ig. G более 70 г/л, Ig. A более 50 г/л, белок Бенс-Джонса в моче более 12 г/сутки. Опухолевая масса более 1, 2 кг/м 2 – высокая (1012 клеток составляют 1 кг опухоли). В каждой стадии выделяют подстадии А (нормальный креатинин сыворотки) и В (повышенный креатинин сыворотки).
: III. Иммунохимическая классификация выделяет варианты множественной миеломы в соответствии с")
Классификация множественной миеломы (4): III. Иммунохимическая классификация выделяет варианты множественной миеломы в соответствии с типом парапротеина сыворотки крови и/или мочи и типом легких цепей. IV. По течению различают следующие формы множественной миеломы: без признаков прогрессирования, медленно прогрессирующая, быстро прогрессирующая.

Вариант формулировки диагноза множественной миеломы подразумевает использование всех перечисленных классификаций. Например: Множественая миелома, диффузно-узловая форма, IIВ стадия, иммунохимический вариант G, тип каппа, медленно прогрессирующее течение.
. 1. Костномозговой синдром. Пролиферирующие в костном мозге миеломные клетки приводят")
Клиника множественной миеломы (1). 1. Костномозговой синдром. Пролиферирующие в костном мозге миеломные клетки приводят к разрушению костного вещества и деструкции костей. Рентгенологически более чем у 80% больных выявляется генерализованный остеопороз с участками остеолиза, патологические переломы костей. Наиболее часто поражаются кости свода черепа, таза, ребер, позвоночника, реже – проксимальные отделы трубчатых костей (плечо, бедро). Особо следует подчеркнуть, что у 1030% больных возможно развитие компрессия спинного мозга или поражение конского хвоста. Это требует быстрой диагностики и лечения (при ламинэктомии в течение первых 24 часов от развития данного осложнения полное восстановление функции спинного мозга отмечается у 50%, частичное – у 30% больных; операция, выполненная позднее, сопровождается резким ухудшением результатов). Трепанобиопсия в 90 -95% случаев выявляет плазмоклеточную пролиферацию с вытеснением нормальных миелоидных элементов.
. 2. Синдром белковой патологии – связан с гипер- и парапротеинемией.")
Клиника множественной миеломы (2). 2. Синдром белковой патологии – связан с гипер- и парапротеинемией. Наиболее частое и серьезное проявление – а) миеломная нефропатия. В основе почечной патологии лежит отложение в канальцах и клубочках имеющегося в избытке моноклонового иммуноглобулина или его дериватов, а также гиперкальциемия. На этой основе развиваются склеротические процессы в почках с прогрессирующей их недостаточностью. Выявляется у половины больных и проявляется упорной протеинурией и постепенно развивающейся хронической почечной недостаточностью (вторая по частоте причина летальных исходов после инфекционных осложнений). У 15% больных миеломной болезнью развивается б) параамилоидоз. В отличие от классического вторичного амилоидоза, при котором страдает печень, селезенка и почки, параамилоидоз поражает органы, богатые коллагеном – мышцы (в том числе сердце, язык), дерму, сухожилия и суставы. Вследствие амилоидной инфильтрации нервов могут развиваться нейропатии. Для диагностики параамилоидоза необходима биопсия кожи, слизистой полости рта, прямой кишки, мышц и т. д.
. 3. Синдром недостаточности антител. Недостаточность продукции нормальных иммуноглобулинов вызвана угнетением")
Клиника множественной миеломы (3). 3. Синдром недостаточности антител. Недостаточность продукции нормальных иммуноглобулинов вызвана угнетением нормального лимфоцитопоэза опухолевыми клетками. Происходит резкое снижение уровня нормальных иммуноглобулинов до полного их исчезновения, а также нарушения клеточного иммунитета. Частые и тяжелые бактериальные инфекции, особенно дыхательных и мочевыводящих путей.
. 4. Синдром висцеральных поражений. Причина этого – специфическая миеломная пролиферация.")
Клиника множественной миеломы (4). 4. Синдром висцеральных поражений. Причина этого – специфическая миеломная пролиферация. Опухолевые плазмоклеточные инфильтраты обнаруживаются практически во всех органах, они могут провоцировать развитие соответствующей патологии, в частности сердечной недостаточности, ревматоидоподобного синдрома и т. п. У 10% больных – гепато- и/или спленомегалия. Висцеральные поражения не имеют большой прогностической значимости.
. 5. Синдром повышенной вязкости (гипервискозный синдром). Избыток белковой продукции может")
Клиника множественной миеломы (5). 5. Синдром повышенной вязкости (гипервискозный синдром). Избыток белковой продукции может приводить к появлению синдрома повышенной вязкости (резкий астенический синдром, парестезии, нарушения кровообращения по микроциркупяторному типу, синдром Рейно, нарушения периферического кровотока вплоть до гангрены дистальных отделов конечностей, тромбозы, инфаркты внутренних органов, геморрагический синдром по типу васкулита – чаще носовые, десневые кровотечения, ретинопатия – вплоть до полной потери зрения). Нарушения микроциркуляции в сосудах головного мозга могут приводить к развитию парапротеинемической комы.
. 6. Гиперкальциемия – при обширном остеолизе, в терминальной стадии болезни,")
Клиника множественной миеломы (6). 6. Гиперкальциемия – при обширном остеолизе, в терминальной стадии болезни, при почечной недостаточности – проявляется тошнотой, рвотой, сонливостью, потерей аппетита и нарушениями ориентации в пространстве.
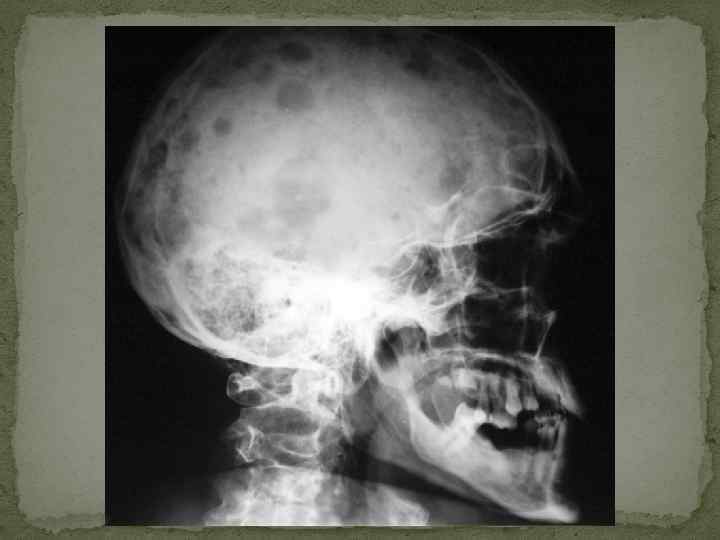
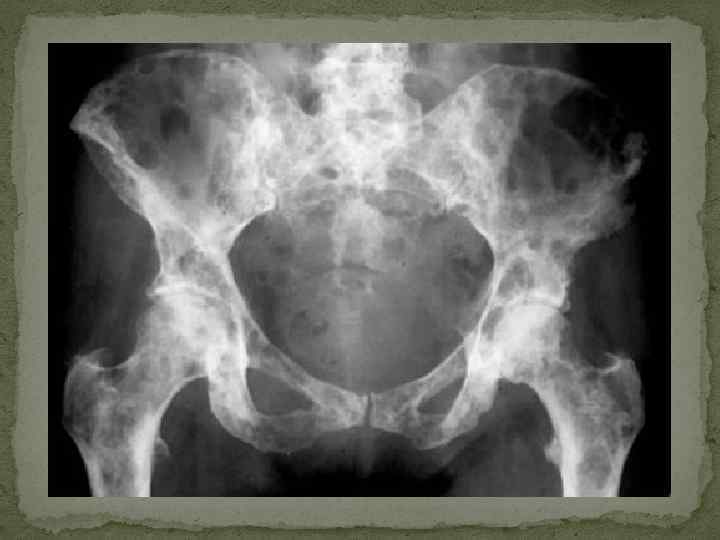
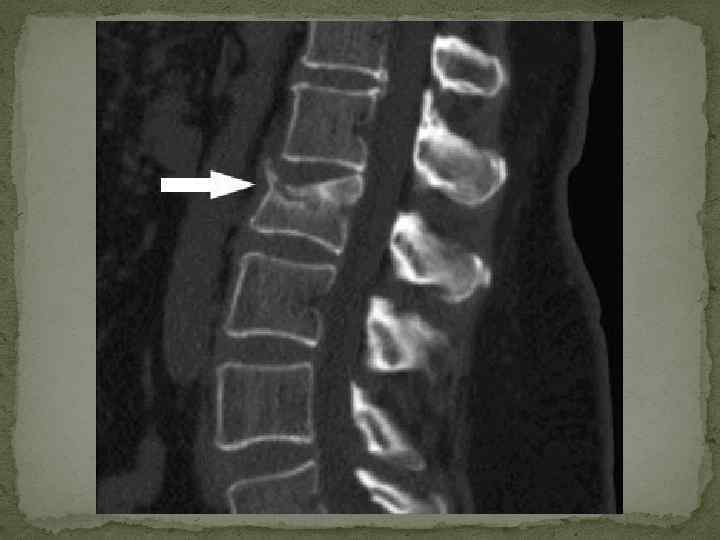
 плазмоцитоз костного мозга (выше")
Диагностика множественной миеломы. Классическая триада при миеломе включает: 1) плазмоцитоз костного мозга (выше 10%); 2) лизис костей с костными болями и 3) наличие М-компонента в сыворотке крови и (или) моче.

ОАК при множественной миеломе Общий анализ крови – нет специфических признаков. Наиболее часто встречаются нормохромная анемия, ускорение СОЭ (до 5080 мм/ч и более), реже отмечаются умеренная лейко- и тромбоцитопения. Необходимо помнить, что при миеломе Бенс. Джонса СОЭ чаще остается в пределах нормы. Эозинофилия – в 30% случаев. Обнаружение плазматических клеток в общем анализе крови не является доказательством миеломной болезни!!!

4, 5 1 0 2 0 4 60 мм/ч 56 30 7

2, 8 2 0 4 0 2 46 28 5 Плазматические клетки 13 72 мм/ч

3, 6 1 0 7 0 2 70 мм/ч 52 34 4

Лабораторные обследования при миеломе При биохимических и иммунологических исследованиях отмечаются гиперпротеинемия, повышение вязкости, парапротеинемия в сыворотке крови более 30 г/л или менее 30 г/л, но сопровождающаяся снижением уровня нормальных иммуноглобулинов. Парапротеины определяются с помощью электрофореза сыворотки крови и/или мочи в виде узкой полосы моноклонового белка в зоне миграции альфа-2 -гамма-глобулинов (М-градиент – графическое изображение белковых фракций). Уточнение характера парапротеина (тип тяжелой и легкой цепи) производится при иммуноэлектрофорезе. Кроме парапротеинемии, отмечается снижение уровня нормальных иммуноглобулинов. Существенное дифференциально-диагностическое и прогностическое значение имеет также определение уровня бета-2 -микроглобулина, несколько меньшее – ЛДГ.

Электрофорез белков

Иммуноэлектрофорез белков

Миелограмма при множественной миеломе Миелограмма – основной метод – большое количество плазматических клеток с ассиметрично расположенным ядром и пенистой цитоплазмой (более 10% – патогномоничный признак). В связи с неравномерностью поражения костного мозга у ряда больных приходится прибегать к повторным стернальным пункциям, трепанобиопсии.

Дообследование при миеломе Рентгенография черепа – симптом пробойника (штампованные дефекты, «кучерявый череп» , «череп в виде шляпы, изъеденной молью» ) – не является основанием для диагноза, а лишь подтверждает его. Исследование мочи – диагностически значим уровень белка Бенс-Джонса более 50 мг/л. Необходимо помнить, что в связи с опасностью развития необратимой почечной недостаточности (образование нерастворимого комплекса парапротеин + йодсодержащее рентгенконтрастное вещество) при миеломе Бенс. Джонса категорически противопоказано проведение внутривенной урографии.
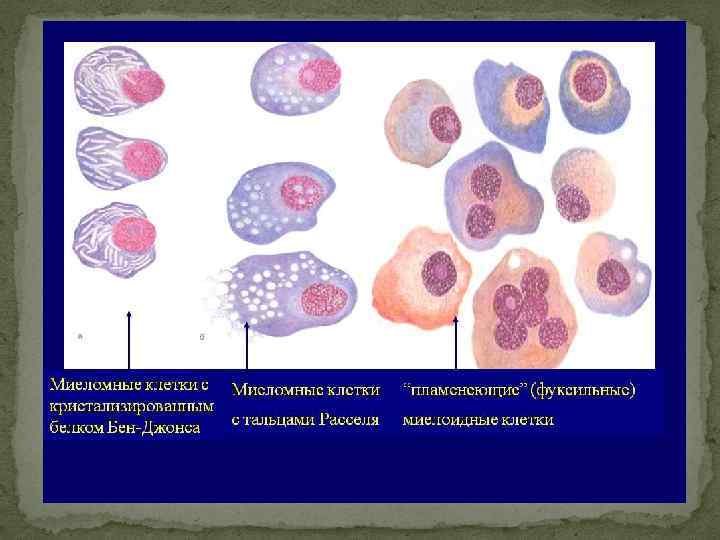
 общие анализы крови и мочи; 2)")
Объем диагностических исследований при множественной миеломе включает: 1) общие анализы крови и мочи; 2) миелограмму (предпочтительно из двух точек) + трепанобиопсию; 3) рентгенографию костей (череп, таз, трубчатые кости); 4) исследование общего белка, определение Мградиента, желательно в сочетании с иммуноэлектрофорезом белков сыворотки крови и/или мочи; 5) исследование функции почек (клиренс креатинина, проба Зимницкого); 6) печеночные тесты; 7) определение уровня кальция сыворотки крови.

В сомнительных случаях, например при несекретирующей миеломе, отсутствии высокого процента плазмоцитов в костном мозге диагноз устанавливают в специализированном учреждении. Подозрительны следующие признаки: немотивированное нарастающее на протяжении ряда лет повышение СОЭ (следствие избытка гаммаглобулинов в крови), упорная протеинурия (не альбуминурия, а глобулинурия, устанавливаемая методом электрофореза мочи), проявление остеодеструктивных очагов, анемия, лейкоцито- и тромбоцитопения. Само по себе установление диагноза миеломной болезни не является показанием к цитостатической терапии; ее назначает специалист. По Харрисону, множественная миелома имеет три диагностических критерия: боли в костях, ускорение СОЭ, плазмоклеточная пролиферация в костном мозге. Эти признаки не являются обязательными у каждого конкретного больного, но наличие данной триады делает диагноз множественной миеломы практически бесспорным.
. При начальной стадии заболевании принята выжидательная тактика – диспансерное наблюдение")
Лечение множественной миеломы (1). При начальной стадии заболевании принята выжидательная тактика – диспансерное наблюдение и принятие решения о начале лечения только при увеличении объема опухоли или ускорении ее роста.
. Трансплантация (пересадка) костного мозга. Единственный метод, позволяющий добиться полного излечения.")
Лечение множественной миеломы (2). Трансплантация (пересадка) костного мозга. Единственный метод, позволяющий добиться полного излечения. Применяется при множественной миеломе довольно редко, так как выполнение трансплантации костного мозга пациентам пожилого возраста сопровождается высоким риском осложнений. Разрабатываются новые методики трансплантации, снижающие риск осложнений.
. Химиотерапия. Основной принцип химиотерапии – быстрое освобождение организма от опухолевых")
Лечение множественной миеломы (3). Химиотерапия. Основной принцип химиотерапии – быстрое освобождение организма от опухолевых клеток с помощью комбинации цитостатических препаратов в достаточных дозах и за определенный отрезок времени. Выбор схемы химиотерапии осуществляется индивидуально. Результаты терапии оцениваются через 3 месяца на основании снижения уровня М-компонента, уменьшения разрушения костей, улучшения состояния больного и лабораторных показателей (улучшение состояния запаздывает по сравнению с лабораторными критериями). При достижении улучшения терапию нужно продолжать длительно – несколько лет, однако это нередко приводит к развитию других злокачественных опухолей.
. Локальная лучевая терапия (воздействие ионизирующего излучения на определенные зоны организма")
Лечение множественной миеломы (4). Локальная лучевая терапия (воздействие ионизирующего излучения на определенные зоны организма с лечебной целью) показана при ограниченных опухолевых узлах в костях и мягких тканях, болях при сдавлении спинного мозга, а также при угрозе переломов костей за счет роста опухоли. Малые дозы облучения с целью «обезболивания» применяться не должны, так как они повышают вероятность рецидива в облученной зоне примерно в 5 раз.
,")
Коррекция нарушений белкового и минерального обмена. Используются анаболические стероиды (препараты, усиливающие синтез нормальных белков), препараты кальцитонина и витамина D (усиливающие поступление кальция из крови в клетки), бифосфонаты (препараты, улучшающие восстановление костей). Все эти препараты применяются после стойкого ответа на химиотерапию.

Лечение инфекционных осложнений. Применяется комбинация антибиотиков, противогрибковые и противовирусные препараты и др.

Лечение почечной недостаточности. Включает: o диета с ограничением белка, o обильное питье, o при задержке жидкости – диуретики (мочегонные препараты), o противоазотемические препараты (снижающие уровень креатинина – продукта распада белка), o энтеросорбенты (препятствующие всасыванию из кишечника вредных веществ). Хроническая почечная недостаточность, существующая более одного- полутора месяцев, требует перевода больных на экстракорпоральные методы лечения (очистку крови за пределами организма с помощью специальных приборов) и в перспективе – на трансплантацию (пересадку) почки.

Экстракорпоральные методы лечения (то есть методы очистки крови за пределами организма при помощи специальных приборов) – повторные процедуры плазмафереза, гемосорбции, гемодиализ – способствуют удалению из организма избытка белка, снижают риск возникновения кровотечений и почечной недостаточности, препятствуют развитию парапротеинемической комы (утрата сознания с отсутствием реакции на внешние раздражители при закрытии просвета сосудов мозга белком).

Хирургическое лечение. Используются при переломах костей и сдавлении спинного мозга опухолью.

Переливание эритроцитарной массы Выполняется при развитии анемии (снижения уровня гемоглобина – особого вещества эритроцитов – красных клеток крови – переносящего кислород) за счет подавления опухолью в костном мозге нормального образования эритроцитов. Проводится по жизненным показаниям (то есть при наличии угрозы жизни пациенту). Угрозой жизни пациенту с анемией являются два состояния: o Анемическая кома (утрата сознания с отсутствием реакции на внешние раздражители вследствие недостаточного поступления кислорода к головному мозгу в результате значительного или быстро развившегося снижения количества эритроцитов); o Тяжелая степень анемии (то есть уровень гемоглобина крови ниже 70 г/л).

Лечебная физкультура и максимально возможная индивидуальная физическая нагрузка.

Осложнения множественной миеломы. Переломы костей за счет роста в них опухоли. Сдавление спинного мозга опухолью с нарушением чувствительности и движений в конечностях. Инфекционные осложнения (органов дыхания, почек и др. ). Это основная причина смерти больных множественной миеломой. Анемия. Повышенная кровоточивость. Парапротеинемическая кома (утрата сознания и реакции на внешние раздражители) за счет закрытия просвета сосудов головного мозга белком. Хроническая почечная недостаточность.

Длительность жизни при множественной миеломе при использовании современного лечения составляет от нескольких месяцев до 10 лет (в среднем 24 -30 месяцев).

Профилактика хронических лейкозов не разработана, так как отсутствуют факторы риска, на которые можно повлиять, а также отсутствуют методы выявления этого заболевания до появления симптомов.

Литература Longo L. D. Harrison's Hematology and Oncology. Mc. Graw-Hill Medical, 2010, 768 p. Абдулкадыров К. М. Гематология. М. : ЭКСМО, СПб. : Сова, 2004. – 928 с. Алексеев Н. А. Анемии. СПб. : Гиппократ, 2004. — 512 с. Альпидовский В. К. и др. Миелопролиферативные заболевания. Москва: РУДН, 2012. — 32 с. Андерсон Ш. , Поулсен К. Атлас гематологии. М. : Логосфера, 2007. — 608 с. Булатов В. П. , Черезова И. Н. и др. Гематология детского возраста. 2 -е изд. , доп. и перераб. – Казань: КГМУ, 2005. – 176 c. Воробьев А. И. (ред. ). Руководство по гематологии. Том 3. М. : Ньюдиамед, 2005. - 416 с. Дроздова М. В. Заболевания крови. СПб, Звезда, 2009. – 408 с. Кобец Т. В. , Бассалыго Г. А. Курс лекций по детской гематологии. Симферополь: КМУ им. С. И. Георгиевского, 2000. – 77 с. Козинец Г. И. (ред. ) Практическая трансфузиология. М. : Практическая медицина, 2005. – 544 с. Кузнецова Е. Ю. , Тимофеева Л. Н. (сост. ) Внутренние болезни: гематология. Красноярск: Крас. ГМУ, 2010. — 114 с. Луговская С. А. , Почтарь М. Е. Гематологический атлас. Тверь: Триада, 2004. – 242 с. Мамаев Н. Н. Гематология: руководство для врачей. СПб. : Спец. Лит, 2008. – 543 с. Основы клинической гематологии. Ермолов С. Ю. , Курдыбайло Ф. В. , Радченко В. Г. , Рукавицын О. А. , Шилова Е. Р. – Под ред. Радченко В. Г. Справочное пособие. — М. : Диалект, 2003. – 304 с. Соловьев А. В, Ракита Д. Р. Гематология. Рязань: Ряз. ГМУ, 2010. — 120 с. Телевная Л. Г. , Грицаева Т. Ф. и др. Интерпретация результатов автоматизированного гематологического анализа (клиниколабораторные аспекты). Омск: ОГМА, 2008. — 37 с. Тимофеева Л. Н. Клинические синдромы в гематологии. Методические рекомендации. – Красноярск: Крас. ГМА, 2006. – 38 с. http: //www. eurolab. ua/diseases/760 http: //vse-zabolevaniya. ru/bolezni-onkologii/mnozhestvennaja-mieloma. html http: //www. likar. info/azbuka-zdorovya/article-56115 -mnozhestvennaya-mieloma/ http: //www. pror. ru/forms_big_myeloma. shtml

Спасибо за внимание!
Лейкозы.pptx