

480f5efd00714d526e32b49214ed82a7.ppt
- Количество слайдов: 28
 LECTURE 91 -15 Analysis of Stage-Specific Gene Expression : Expression Sequence Tags Petrus Tang, Ph. D. Graduate Institute of Basic Medical Sciences and Bioinformatics Center, Chang Gung University. petang@mail. cgu. edu. tw http: //petang. cgu. edu. tw 27 th December 2002
LECTURE 91 -15 Analysis of Stage-Specific Gene Expression : Expression Sequence Tags Petrus Tang, Ph. D. Graduate Institute of Basic Medical Sciences and Bioinformatics Center, Chang Gung University. petang@mail. cgu. edu. tw http: //petang. cgu. edu. tw 27 th December 2002
 Prokaryotic Ongoing") THE WORLD OF GENOMICS Published Complete Genome Projects: 95 (including 3 chromosomes) Prokaryotic Ongoing Genome Projects: 310 Eukaryotic Ongoing Genome Projects: 211 (including 11 chromosomes) Last update: 18 July 2002
THE WORLD OF GENOMICS Published Complete Genome Projects: 95 (including 3 chromosomes) Prokaryotic Ongoing Genome Projects: 310 Eukaryotic Ongoing Genome Projects: 211 (including 11 chromosomes) Last update: 18 July 2002
 Gen. Bank Sequences Gen. Bank® is the National Institute of Health genetic sequence database, an annotated collection of all publicly available DNA sequences. There approximately 20, 648, 748, 345 bases in 17, 471, 130 sequence records as of June 2002 R 130 (12, 055, 326 sequences in d. BEST, 4. 500, 000 from Homo sapiens).
Gen. Bank Sequences Gen. Bank® is the National Institute of Health genetic sequence database, an annotated collection of all publicly available DNA sequences. There approximately 20, 648, 748, 345 bases in 17, 471, 130 sequence records as of June 2002 R 130 (12, 055, 326 sequences in d. BEST, 4. 500, 000 from Homo sapiens).
 are developed") High Throughput Technologies: The future of Molecular Medicine High Throughput Technologies (HTTs) are developed to produce huge amount of information from genome projects, but they have clear potential in mass screening and diagnostics of Infectious Diseases. The application of HTTs may revolutionize diagnostic techniques and replacing multiple individual assays. Genome Transcriptome Proteome m. RNA Gene Protein Gene Products
High Throughput Technologies: The future of Molecular Medicine High Throughput Technologies (HTTs) are developed to produce huge amount of information from genome projects, but they have clear potential in mass screening and diagnostics of Infectious Diseases. The application of HTTs may revolutionize diagnostic techniques and replacing multiple individual assays. Genome Transcriptome Proteome m. RNA Gene Protein Gene Products
 Gene Expression & Post-Translational Modification of Proteins Muscle cell Skin cell Gene A Gene B Gene C Cell Growth, External Stress Gene A Gene B Gene C Nerve cell Normal cell Cancer cell
Gene Expression & Post-Translational Modification of Proteins Muscle cell Skin cell Gene A Gene B Gene C Cell Growth, External Stress Gene A Gene B Gene C Nerve cell Normal cell Cancer cell
 Analysis of Stage-Specific Gene Expression Northern Hybridization RT-PCR Differential Display, Subtraction Library, Serial Analysis of Gene Expression (SAGE) Expressed Sequence Tags (EST) Real-Time PCR Microarry Analysis of 10, 000 -50, 000 messages in a transcriptome will generate a relevant profile of gene expression within a cell, providing a quantitative measurement of transcripts for gene discovery.
Analysis of Stage-Specific Gene Expression Northern Hybridization RT-PCR Differential Display, Subtraction Library, Serial Analysis of Gene Expression (SAGE) Expressed Sequence Tags (EST) Real-Time PCR Microarry Analysis of 10, 000 -50, 000 messages in a transcriptome will generate a relevant profile of gene expression within a cell, providing a quantitative measurement of transcripts for gene discovery.
 Microarray 10, 000 Clones per slide
Microarray 10, 000 Clones per slide
 1. Mix 5 µg total RNA with oligo") Serial Analysis of Gene Expression (SAGE) 1. Mix 5 µg total RNA with oligo d. T magnetic beads 2. Synthesize double-strand c. DNA 3. Digest with Nla. III to form one end of the tag 4. Divide in half and ligate 40 bp adapters (A and B) containing the recognition sequence for the type. II restriction enzyme Bsm. F 1 5. Cleave with Bsm. F 1 to form ~ 50 bp tag (40 bp adaptor/13 bp tag) 6. Fill in 5' overhangs and ligate to form a ~ 100 bp ditag 7. PCR amplify using ditag primers 1 and 2 8. Cut 40 bp adapters with Nla III to release the 26 bp ditag 9. Ligate ditags to form concatemers 10. Clone and sequence
Serial Analysis of Gene Expression (SAGE) 1. Mix 5 µg total RNA with oligo d. T magnetic beads 2. Synthesize double-strand c. DNA 3. Digest with Nla. III to form one end of the tag 4. Divide in half and ligate 40 bp adapters (A and B) containing the recognition sequence for the type. II restriction enzyme Bsm. F 1 5. Cleave with Bsm. F 1 to form ~ 50 bp tag (40 bp adaptor/13 bp tag) 6. Fill in 5' overhangs and ligate to form a ~ 100 bp ditag 7. PCR amplify using ditag primers 1 and 2 8. Cut 40 bp adapters with Nla III to release the 26 bp ditag 9. Ligate ditags to form concatemers 10. Clone and sequence
 What are ESTs? Expressed Sequence Tags are small pieces of DNA sequence (usually 200 to 500 nucleotides long) that are generated by sequencing either one or both ends of an expressed gene. The idea is to sequence bits of DNA that represent genes expressed in certain cells, tissues, or organs from different organisms and use these "tags" to fish a gene out of a portion of chromosomal DNA by matching base pairs. The challenge associated with identifying genes from genomic sequences varies among organisms and is dependent upon genome size as well as the presence or absence of introns--the intervening DNA sequences interrupting the protein coding sequence of a gene.
What are ESTs? Expressed Sequence Tags are small pieces of DNA sequence (usually 200 to 500 nucleotides long) that are generated by sequencing either one or both ends of an expressed gene. The idea is to sequence bits of DNA that represent genes expressed in certain cells, tissues, or organs from different organisms and use these "tags" to fish a gene out of a portion of chromosomal DNA by matching base pairs. The challenge associated with identifying genes from genomic sequences varies among organisms and is dependent upon genome size as well as the presence or absence of introns--the intervening DNA sequences interrupting the protein coding sequence of a gene.
 5’-EST 5` Coding Sequence (CDS) * START 5`-Untranlasted region (UTR)") Expressed Sequence Tags (EST) 5’-EST 5` Coding Sequence (CDS) * START 5`-Untranlasted region (UTR) 3`-UTR * STOP AAAAAA 3’-EST 3`
Expressed Sequence Tags (EST) 5’-EST 5` Coding Sequence (CDS) * START 5`-Untranlasted region (UTR) 3`-UTR * STOP AAAAAA 3’-EST 3`
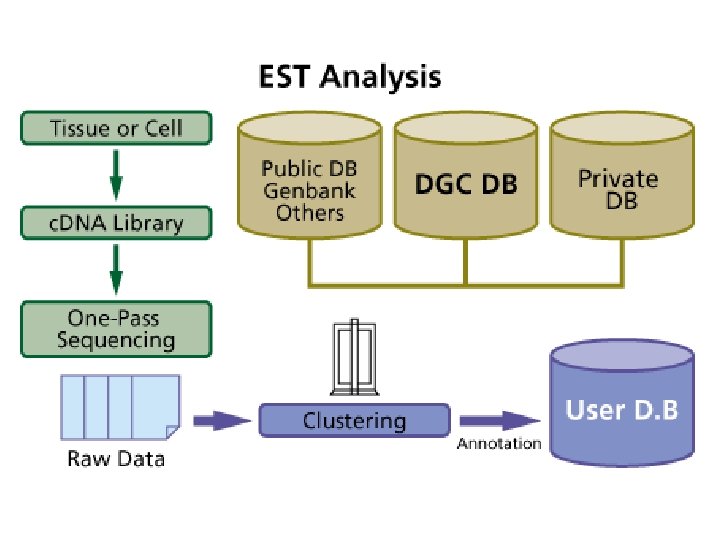
 Basic Features and Tools of an Automated EST Analysis Pipeline ▲ Relational database (Oracle 8 i) ▲ Automatic data validation ▲ Quality score generation ▲ Automatic trimming of low-quality, vector, adaptor, poly-A tails, low-complexity and contaminant sequences ▲ Automatic running of selected blast algorithms, with user-defined parameters, user selected reference databases, and storage of top results (by userdefined cutoffs) in the database ▲ Includes a web interface for viewing the data in the database, according to the permissions allowed to the viewer (by individual, project, lab or institution) ▲ Includes a Java tool for db. EST submission of newly generated ESTs at intervals define by the users ▲ System can be readily and simply deployed at any of the partner's institutions ▲ Includes methods for defining a Unigene set for a library. Additional functionalities are needed by the members of the current co -development group, including: ▲ Tissue or organism, integration of gene expression data. ▲ Annotations: Gene ontology annotations, functional motif annotation, metabolic
Basic Features and Tools of an Automated EST Analysis Pipeline ▲ Relational database (Oracle 8 i) ▲ Automatic data validation ▲ Quality score generation ▲ Automatic trimming of low-quality, vector, adaptor, poly-A tails, low-complexity and contaminant sequences ▲ Automatic running of selected blast algorithms, with user-defined parameters, user selected reference databases, and storage of top results (by userdefined cutoffs) in the database ▲ Includes a web interface for viewing the data in the database, according to the permissions allowed to the viewer (by individual, project, lab or institution) ▲ Includes a Java tool for db. EST submission of newly generated ESTs at intervals define by the users ▲ System can be readily and simply deployed at any of the partner's institutions ▲ Includes methods for defining a Unigene set for a library. Additional functionalities are needed by the members of the current co -development group, including: ▲ Tissue or organism, integration of gene expression data. ▲ Annotations: Gene ontology annotations, functional motif annotation, metabolic
 Data Processing – Raw Nucleotide Sequence EST or SAGE clones sequenced Mega. BRACE 1000 PC Chromas sequence High quality Poor quality Abi format UNIX sequence High quality Poor quality Fasta format Remove uncalled/miscalled bases & vector sequence PHRED algorithm Ewing B et al. (1988) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8(3): 175 -85 Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8(3): 186 -94
Data Processing – Raw Nucleotide Sequence EST or SAGE clones sequenced Mega. BRACE 1000 PC Chromas sequence High quality Poor quality Abi format UNIX sequence High quality Poor quality Fasta format Remove uncalled/miscalled bases & vector sequence PHRED algorithm Ewing B et al. (1988) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8(3): 175 -85 Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8(3): 186 -94
 FREEWARES Trace Viewers: In order to take a look at the SCF file you first have to choose a program. Viewers: Very commonly used programs for viewing the sequencing data are CHROMAS (for PC/Windows), Trace. Viewer (for MAC) and Trev (contained in the Gap 4 Database Viewer, for UNIX). DL Seq. Verter™ is a free sequence file format conversion utility by Gene. Studio, Inc. Seq. Verter encapsulates a small subset of the features offered by the Gene. Studio Pro suite of programs. While the standalone Seq. Verter is a simple dialog-based utility, the free Seq. Verter component of the Gene. Studio suite adds sophisticated viewers and sequence formatting functions, including a viewer for automatic DNA sequencer chromatogram files (traces). http: //www. genestudio. com/seqverter. htm DL Octopus is an interactive program designed for the rapid interpretation of BLAST, BLAST -2 and FASTA output text files. It provides an easy-to-use graphical user interface for both experienced and inexperienced users with sequence comparison analysis based on the widely-used BLAST serie of softwares and FASTA. Octopus is able to read results files coming from various BLAST and BLAST 2 servers, the GCG's BLAST and the original FASTA 3 program. DL
FREEWARES Trace Viewers: In order to take a look at the SCF file you first have to choose a program. Viewers: Very commonly used programs for viewing the sequencing data are CHROMAS (for PC/Windows), Trace. Viewer (for MAC) and Trev (contained in the Gap 4 Database Viewer, for UNIX). DL Seq. Verter™ is a free sequence file format conversion utility by Gene. Studio, Inc. Seq. Verter encapsulates a small subset of the features offered by the Gene. Studio Pro suite of programs. While the standalone Seq. Verter is a simple dialog-based utility, the free Seq. Verter component of the Gene. Studio suite adds sophisticated viewers and sequence formatting functions, including a viewer for automatic DNA sequencer chromatogram files (traces). http: //www. genestudio. com/seqverter. htm DL Octopus is an interactive program designed for the rapid interpretation of BLAST, BLAST -2 and FASTA output text files. It provides an easy-to-use graphical user interface for both experienced and inexperienced users with sequence comparison analysis based on the widely-used BLAST serie of softwares and FASTA. Octopus is able to read results files coming from various BLAST and BLAST 2 servers, the GCG's BLAST and the original FASTA 3 program. DL
 EST Analysis : Clustering ALGORITHM FUNCTION PHRED Remove uncalled/miscalled bases & vector sequence PHRAP Assemble clones to from contigs CONSED Contig viewer & screen for misassemblies Wu-Blastn Group contigs to form clusters of related contigs Blastx Homology search against self-generated dbases CONTIGS Clusters Singletons
EST Analysis : Clustering ALGORITHM FUNCTION PHRED Remove uncalled/miscalled bases & vector sequence PHRAP Assemble clones to from contigs CONSED Contig viewer & screen for misassemblies Wu-Blastn Group contigs to form clusters of related contigs Blastx Homology search against self-generated dbases CONTIGS Clusters Singletons
 Similarity Search: Blastx BLAST uses a heuristic algorithm which seeks local as opposed to global alignments and is therefore able to detect relationships among sequences which share only isolated regions of similarity (Altschul et al. , 1990) Nucleotide query translated to six reading frames vs protein database TV 007 D 02 WWW Blastx-nr Blastx-pfam, smart GCG Blastx-GCG format Blastx-Octopus viewer
Similarity Search: Blastx BLAST uses a heuristic algorithm which seeks local as opposed to global alignments and is therefore able to detect relationships among sequences which share only isolated regions of similarity (Altschul et al. , 1990) Nucleotide query translated to six reading frames vs protein database TV 007 D 02 WWW Blastx-nr Blastx-pfam, smart GCG Blastx-GCG format Blastx-Octopus viewer
 Inter. Pro provides an integrated view of the commonly used signature databases, and has an intuitive interface for text- and sequence-based searches. Bioinformatics infrastructural activities are crucial to modern biological research. Complete and up-to-date databases of biological knowledge are vital for the increasingly information-dependent biological and biotechnological research. Secondary protein databases on functional sites and domains like PROSITE, PRINTS, SMART, Pfam, Pro. Dom, etc. are vital resources for identifying distant relationships in novel sequences, and hence for predicting protein function and structure. Unfortunately, these signature databases do not share the same formats and nomenclature, and each database has is own strengths and weaknesses. To capitalise on these, the following partners: EBI, SIB, University of Manchester, Sanger Institute, GENE-IT, CNRS/INRA, LION bioscience AG and University of Bergen unified PROSITE, PRINTS, Pro. Dom and Pfam into Inter. Pro (Integrated resource of Protein Families, Domains and Sites). The latest databases to join the project were SMART, and more recently, TIGRFAMs.
Inter. Pro provides an integrated view of the commonly used signature databases, and has an intuitive interface for text- and sequence-based searches. Bioinformatics infrastructural activities are crucial to modern biological research. Complete and up-to-date databases of biological knowledge are vital for the increasingly information-dependent biological and biotechnological research. Secondary protein databases on functional sites and domains like PROSITE, PRINTS, SMART, Pfam, Pro. Dom, etc. are vital resources for identifying distant relationships in novel sequences, and hence for predicting protein function and structure. Unfortunately, these signature databases do not share the same formats and nomenclature, and each database has is own strengths and weaknesses. To capitalise on these, the following partners: EBI, SIB, University of Manchester, Sanger Institute, GENE-IT, CNRS/INRA, LION bioscience AG and University of Bergen unified PROSITE, PRINTS, Pro. Dom and Pfam into Inter. Pro (Integrated resource of Protein Families, Domains and Sites). The latest databases to join the project were SMART, and more recently, TIGRFAMs.
 Annotation - GO GENE ONTOLOGYTM CONSORTIUM http: //www. geneontology. org The goal of the Gene Ontology. TM Consortium is to produce a dynamic controlled vocabulary that can be applied to all organisms even as knowledge of gene and protein roles in cells is accumulating and changing. Molecular Function the tasks performed by individual gene products; examples are transcription factor and DNA helicase. Biological Process broad biological goals, such as mitosis or purine metabolism, that are accomplished by ordered assemblies of molecular functions. Cellular Component p 53 subcellular structures, locations, and macromolecular complexes; examples include nucleus, telomere, and origin recognition complex.
Annotation - GO GENE ONTOLOGYTM CONSORTIUM http: //www. geneontology. org The goal of the Gene Ontology. TM Consortium is to produce a dynamic controlled vocabulary that can be applied to all organisms even as knowledge of gene and protein roles in cells is accumulating and changing. Molecular Function the tasks performed by individual gene products; examples are transcription factor and DNA helicase. Biological Process broad biological goals, such as mitosis or purine metabolism, that are accomplished by ordered assemblies of molecular functions. Cellular Component p 53 subcellular structures, locations, and macromolecular complexes; examples include nucleus, telomere, and origin recognition complex.
 Kyto Encyclopedia") Classification According to Metabolic & Signalling Pathways Biocarta ( http: //biocarta. com) Kyto Encyclopedia of Genes &Genomes http: //www. genome. ad. jp/kegg/ The Cancer Genome Anatomy Project (CGAP) http: //cgap. nci. nih. gov/
Classification According to Metabolic & Signalling Pathways Biocarta ( http: //biocarta. com) Kyto Encyclopedia of Genes &Genomes http: //www. genome. ad. jp/kegg/ The Cancer Genome Anatomy Project (CGAP) http: //cgap. nci. nih. gov/
 Annotation ESTs are categorized into the following classes: ESTs shows homology to known protein motifs/domains Unique ESTs with no matces ESTs matches exactly to known protein sequences
Annotation ESTs are categorized into the following classes: ESTs shows homology to known protein motifs/domains Unique ESTs with no matces ESTs matches exactly to known protein sequences
 Cell Component
Cell Component
 Molecular Function
Molecular Function
 Biological Process
Biological Process
 Automated EST Analysis Pipeline Gen. Bank® is the National Institute of Health genetic sequence database, an annotated collection of all publicly available DNA sequences. There approximately 20, 648, 748, 345 bases in 17, 471, 130 sequence records as of June 2002 R 130 (12, 055, 326 sequences in d. BEST, 4. 500, 000 from Homo sapiens). Project Management Sequence Management Clustering Sequence Analysis Annotation d. BEST 12, 261, 869 (Aug, 2002)
Automated EST Analysis Pipeline Gen. Bank® is the National Institute of Health genetic sequence database, an annotated collection of all publicly available DNA sequences. There approximately 20, 648, 748, 345 bases in 17, 471, 130 sequence records as of June 2002 R 130 (12, 055, 326 sequences in d. BEST, 4. 500, 000 from Homo sapiens). Project Management Sequence Management Clustering Sequence Analysis Annotation d. BEST 12, 261, 869 (Aug, 2002)
 EST Databases – d. BEST & UNIGENE db. EST (http: //www. ncbi. nlm. nih. gov/db. EST/index. html) is a division of Gen. Bank that contains sequence data and other information on "single-pass" c. DNA sequences, or Expressed Sequence Tags, from a number of organisms. Uni. Gene (http: //www. ncbi. nlm. nih. gov/entrez/query. fcgi? db=unigene) is an experimental system for automatically partitioning Gen. Bank sequences into a non-redundant set of gene-oriented clusters. Each Uni. Gene cluster contains sequences that represent a unique gene, as well as related information such as the tissue types in which the gene has been expressed and map location.
EST Databases – d. BEST & UNIGENE db. EST (http: //www. ncbi. nlm. nih. gov/db. EST/index. html) is a division of Gen. Bank that contains sequence data and other information on "single-pass" c. DNA sequences, or Expressed Sequence Tags, from a number of organisms. Uni. Gene (http: //www. ncbi. nlm. nih. gov/entrez/query. fcgi? db=unigene) is an experimental system for automatically partitioning Gen. Bank sequences into a non-redundant set of gene-oriented clusters. Each Uni. Gene cluster contains sequences that represent a unique gene, as well as related information such as the tissue types in which the gene has been expressed and map location.
 d. BEST Record NCBI d. BEST Accession numbers for Trichomonas vaginalis ESTs BQ 621379~BQ 621732; BQ 625216~BQ 625229; BQ 640771~BQ 640943 1: BQ 640943. TVEST 017. H 09 Tv 30. . . [gi: 21765401] Taxonomy Entry Created: Jul 8 2002 Last Updated: Jul 15 2002 IDENTIFIERS db. EST Id: EST name: Gen. Bank Acc: Gen. Bank gi: 12791004 TVEST 017. H 09 BQ 640943 21765401 CLONE INFO Clone Id: DNA type: (5') c. DNA PRIMERS PCR forward: PCR backward: Sequencing: Poly. A Tail: T 7 T 3 Unknown SEQUENCE ATTACAGCAATTGCCGATGATTGGCATCACTGGCGTATCGAAAACTTTAAG CTCGTTAAAGTTGCAGAGATGGGCGCCTTCCACACAGGAGATTCTTATTTGTATCTTCAC GCTTACCTTGNTTGGCACAAGCTCGTCCATCGTGATATTTACTTCTGGCAGGGCTC CACATCCACAACAGATGAGCGCGGTGCTGTTGCTATCAAGGCTGTTGAACTTGATGACAG ATTTGGAGGCTCTCCAAAGCAACACAGAGAAGTCCAGAACCACGAGTCAGACCAGTTCAT TGGACTCTTCGATCAGTTTGGCGGTGTTCGCTACCTCGATGGCGGTGTTGAATCAGGATT CCACAAAGTCACAACATCTGCAAAGGTTGAGATGTACAGAATCAAGGGAAGAAAGCGCCC AATTCTCCAGATCGTTCCAGCTCAGCGCTCCTCAACCATGGAGATGTTTTCATTAT CCATGC http: //www. ncbi. nlm. nih. gov/db. EST/index. html trichomonas vaginalis AND gbdiv_est[PROP] PUTATIVE ID Assigned by submitter ACTIN-BINDING PROTEIN FRAGMIN P. LIBRARY Lib Name: Tv 30236_PT c. DNA Library Organism: Trichomonas vaginalis Cell line: ATCC 30236 Develop. stage: Trophozoites at mid-log phase Lab host: XL 1 Blue-MRF' Vector: Lambda ZAP-Express (Stratagene) R. Site 1: Eco. RI R. Site 2: Xho. I SUBMITTER Name: Tang, P. Lab: Molecular Regulation and Bioinformatics Laboratory, College of Medicine Institution: Chang Gung University Address: 259 Wenhwa 1 st. Road, Kweishan, Taoyuan 333, Taiwan Tel: +886 3 3283016 EXT 5136 Fax: +886 3 3283031 E-mail: petang@mail. cgu. edu. tw CITATIONS Title: Analysis of Gene Expression Profile in Trichomonas vaginalis by EST Sequencing Authors: Zhou, Y. , Shu, W. M. , Huang, S. C. C. , Huang, K. Y. , Tang, P. Year: 2003 Status: Unpublished
d. BEST Record NCBI d. BEST Accession numbers for Trichomonas vaginalis ESTs BQ 621379~BQ 621732; BQ 625216~BQ 625229; BQ 640771~BQ 640943 1: BQ 640943. TVEST 017. H 09 Tv 30. . . [gi: 21765401] Taxonomy Entry Created: Jul 8 2002 Last Updated: Jul 15 2002 IDENTIFIERS db. EST Id: EST name: Gen. Bank Acc: Gen. Bank gi: 12791004 TVEST 017. H 09 BQ 640943 21765401 CLONE INFO Clone Id: DNA type: (5') c. DNA PRIMERS PCR forward: PCR backward: Sequencing: Poly. A Tail: T 7 T 3 Unknown SEQUENCE ATTACAGCAATTGCCGATGATTGGCATCACTGGCGTATCGAAAACTTTAAG CTCGTTAAAGTTGCAGAGATGGGCGCCTTCCACACAGGAGATTCTTATTTGTATCTTCAC GCTTACCTTGNTTGGCACAAGCTCGTCCATCGTGATATTTACTTCTGGCAGGGCTC CACATCCACAACAGATGAGCGCGGTGCTGTTGCTATCAAGGCTGTTGAACTTGATGACAG ATTTGGAGGCTCTCCAAAGCAACACAGAGAAGTCCAGAACCACGAGTCAGACCAGTTCAT TGGACTCTTCGATCAGTTTGGCGGTGTTCGCTACCTCGATGGCGGTGTTGAATCAGGATT CCACAAAGTCACAACATCTGCAAAGGTTGAGATGTACAGAATCAAGGGAAGAAAGCGCCC AATTCTCCAGATCGTTCCAGCTCAGCGCTCCTCAACCATGGAGATGTTTTCATTAT CCATGC http: //www. ncbi. nlm. nih. gov/db. EST/index. html trichomonas vaginalis AND gbdiv_est[PROP] PUTATIVE ID Assigned by submitter ACTIN-BINDING PROTEIN FRAGMIN P. LIBRARY Lib Name: Tv 30236_PT c. DNA Library Organism: Trichomonas vaginalis Cell line: ATCC 30236 Develop. stage: Trophozoites at mid-log phase Lab host: XL 1 Blue-MRF' Vector: Lambda ZAP-Express (Stratagene) R. Site 1: Eco. RI R. Site 2: Xho. I SUBMITTER Name: Tang, P. Lab: Molecular Regulation and Bioinformatics Laboratory, College of Medicine Institution: Chang Gung University Address: 259 Wenhwa 1 st. Road, Kweishan, Taoyuan 333, Taiwan Tel: +886 3 3283016 EXT 5136 Fax: +886 3 3283031 E-mail: petang@mail. cgu. edu. tw CITATIONS Title: Analysis of Gene Expression Profile in Trichomonas vaginalis by EST Sequencing Authors: Zhou, Y. , Shu, W. M. , Huang, S. C. C. , Huang, K. Y. , Tang, P. Year: 2003 Status: Unpublished
 EST & SAGE Based Microarray Not Pre-selected Can identify Gene Families Real Gene Expressed Products c. DNA vs c. DNA Abundance = Expression Level Normal, U 1, U 2, U 3, U 4, Prognosis, Drug Resistant Bladder Tissue, Normal Bladder Tissue, Cancer Genes m. RNAs Genes c. DNA ESTs Bladder Carcinoma-Specific Microarrays
EST & SAGE Based Microarray Not Pre-selected Can identify Gene Families Real Gene Expressed Products c. DNA vs c. DNA Abundance = Expression Level Normal, U 1, U 2, U 3, U 4, Prognosis, Drug Resistant Bladder Tissue, Normal Bladder Tissue, Cancer Genes m. RNAs Genes c. DNA ESTs Bladder Carcinoma-Specific Microarrays