

курс_2012 функциональные производные часть 1.ppt
- Количество слайдов: 187
 КИСЛОРОДСОДЕРЖАЩИЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ Кислород – элемент VI группы 2 -го периода Периодической системы элементов; порядковый номер 8; атомная масса 16; электроотрицательность 3, 5. Электронная конфигурация в основном состоянии 1 s 22 p 4. На 2 энергетическом уровне в атоме кислорода находятся 2 неспаренных электрона, за счет которых он может образовывать ковалентные связи с другими атомами, и 2 неподеленные электронные пары, которые могут участвовать в образовании донорно-акцепторных связей или вступать в n- *сопряжение с углеводородным фрагментом молекулы.
КИСЛОРОДСОДЕРЖАЩИЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ Кислород – элемент VI группы 2 -го периода Периодической системы элементов; порядковый номер 8; атомная масса 16; электроотрицательность 3, 5. Электронная конфигурация в основном состоянии 1 s 22 p 4. На 2 энергетическом уровне в атоме кислорода находятся 2 неспаренных электрона, за счет которых он может образовывать ковалентные связи с другими атомами, и 2 неподеленные электронные пары, которые могут участвовать в образовании донорно-акцепторных связей или вступать в n- *сопряжение с углеводородным фрагментом молекулы.
 ГИДРОКСИЛПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ Содержат в своем составе ковалентно-связанную группу –ОН Номенклатура и классификация 1. По строению заместителей, связанных с атомом кислорода, различают: - предельные спирты: СН 3 -ОН метиловый спирт, метанол; СН 3 -СН 2 -ОН этиловый спирт, этанол; СН 3 -СН 2 -ОН пропиловый спирт, пропанол-1; СН 3 -СН(ОН)-СН 3 изопропиловый спирт, пропанол-2; - непредельные спирты: СН 2=СН-ОН виниловый спирт (неустойчив); СН 2=СН-СН 2 -ОН аллиловый спирт; - арилсодержащие неароматические спирты: 1. C 6 H 5 CH 2–OH бензиловый спирт; 2. C 6 H 5 -CH(OH)-C 6 H 5 гидроксидифенилметан; 3. C 6 H 5 -CH(OH)-CH 3 -гидроксиэтилбензол;
ГИДРОКСИЛПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ Содержат в своем составе ковалентно-связанную группу –ОН Номенклатура и классификация 1. По строению заместителей, связанных с атомом кислорода, различают: - предельные спирты: СН 3 -ОН метиловый спирт, метанол; СН 3 -СН 2 -ОН этиловый спирт, этанол; СН 3 -СН 2 -ОН пропиловый спирт, пропанол-1; СН 3 -СН(ОН)-СН 3 изопропиловый спирт, пропанол-2; - непредельные спирты: СН 2=СН-ОН виниловый спирт (неустойчив); СН 2=СН-СН 2 -ОН аллиловый спирт; - арилсодержащие неароматические спирты: 1. C 6 H 5 CH 2–OH бензиловый спирт; 2. C 6 H 5 -CH(OH)-C 6 H 5 гидроксидифенилметан; 3. C 6 H 5 -CH(OH)-CH 3 -гидроксиэтилбензол;
. Фенолы – гидроксисоединения, в молекулах которых ОНгруппы связаны непосредственно с") - фенолы (ароматические спирты). Фенолы – гидроксисоединения, в молекулах которых ОНгруппы связаны непосредственно с бензольным ядром 2. По числу гидроксильных групп различают: - одноатомные спирты и фенолы (все вышеперечисленные соединения); - многоатомные спирты и фенолы: НО-СН 2 -ОН этиленгликоль, 1, 2 -этандиол; НО-СН 2 -СН(ОН)-С 2 -ОН глицерин, 1, 2, 3 -пропантриол;
- фенолы (ароматические спирты). Фенолы – гидроксисоединения, в молекулах которых ОНгруппы связаны непосредственно с бензольным ядром 2. По числу гидроксильных групп различают: - одноатомные спирты и фенолы (все вышеперечисленные соединения); - многоатомные спирты и фенолы: НО-СН 2 -ОН этиленгликоль, 1, 2 -этандиол; НО-СН 2 -СН(ОН)-С 2 -ОН глицерин, 1, 2, 3 -пропантриол;
2 неустойчивы") Двухатомные спирты с двумя ОН-группами при одном и том же атоме углерода R-CH(OH)2 неустойчивы и, отщепляя воду, превращаются в альдегиды или кетоны. Спирты R-С(ОН)3 не существуют.
Двухатомные спирты с двумя ОН-группами при одном и том же атоме углерода R-CH(OH)2 неустойчивы и, отщепляя воду, превращаются в альдегиды или кетоны. Спирты R-С(ОН)3 не существуют.
") 3. В зависимости от того, с каким атомом углерода (первичным, вторичным или третичным) связана гидроксигруппа, различают спирты: - первичные R–CH 2–OH; - вторичные R 2 CH–OH; - третичные R 3 C–OH. По номенклатуре ИЮПАК спирты называют, добавляя суффикс –ол к названию соответствующего углеводорода и указывая цифрой положение гидроксигруппы или обозначая группу –ОН как «гидрокси» : пентанол-2 2, 4, 4 -триметилпентанол-2 гидроксиметилциклобутан
3. В зависимости от того, с каким атомом углерода (первичным, вторичным или третичным) связана гидроксигруппа, различают спирты: - первичные R–CH 2–OH; - вторичные R 2 CH–OH; - третичные R 3 C–OH. По номенклатуре ИЮПАК спирты называют, добавляя суффикс –ол к названию соответствующего углеводорода и указывая цифрой положение гидроксигруппы или обозначая группу –ОН как «гидрокси» : пентанол-2 2, 4, 4 -триметилпентанол-2 гидроксиметилциклобутан
 Спирты R-OH также называют по алкильному фрагменту: н-бутиловый спирт изо-бутиловый спирт втор-бутиловый спирт трет-бутиловый спирт По устаревшей карбинольной номенклатуре спирты называют как производные первого члена гомологического ряда – карбинола CH 3 OH: дифенилкарбинол метилфеникарбинол
Спирты R-OH также называют по алкильному фрагменту: н-бутиловый спирт изо-бутиловый спирт втор-бутиловый спирт трет-бутиловый спирт По устаревшей карбинольной номенклатуре спирты называют как производные первого члена гомологического ряда – карбинола CH 3 OH: дифенилкарбинол метилфеникарбинол
 Строение функциональной группы О–Н и С–О - ковалентные полярные связи. Электронная плотность на обеих связях смещена к более электроотрицательному атому кислорода: 110 о Дипольный момент связи С–О составляет 0, 70 D, а связи О–Н – 1, 51 D. Разрыв таких связей происходит преимущественно гетеролитически (по ионному механизму). Энергии связей С-О и О-Н в спиртах равны соответственно 350 и 460 к. Дж/моль, угол СО-Н составляет порядка 110 о в зависимости от структуры углеводородного заместителя.
Строение функциональной группы О–Н и С–О - ковалентные полярные связи. Электронная плотность на обеих связях смещена к более электроотрицательному атому кислорода: 110 о Дипольный момент связи С–О составляет 0, 70 D, а связи О–Н – 1, 51 D. Разрыв таких связей происходит преимущественно гетеролитически (по ионному механизму). Энергии связей С-О и О-Н в спиртах равны соответственно 350 и 460 к. Дж/моль, угол СО-Н составляет порядка 110 о в зависимости от структуры углеводородного заместителя.
 Из двух неподленных электронных пар одна находится на s-орбитали, другая – на pz-орбитали. s. Электронная пара может образовывать связи с частицами, имеющими вакантные орбитали, поэтому спирты при взаимодействии, например, с кислотами, легко протонируются. В результате большей электроотрицательности атома кислорода гидроксигруппа проявляет отрицательный индукционный эффект (-I-эффект) по отношению к углеводородному заместителю и в спиртах выступает как электроноакцептор. Спирты имеют большое сходство с водой, но алкильный заместитель увеличивает ковалентность связи ОН: R О-Н.
Из двух неподленных электронных пар одна находится на s-орбитали, другая – на pz-орбитали. s. Электронная пара может образовывать связи с частицами, имеющими вакантные орбитали, поэтому спирты при взаимодействии, например, с кислотами, легко протонируются. В результате большей электроотрицательности атома кислорода гидроксигруппа проявляет отрицательный индукционный эффект (-I-эффект) по отношению к углеводородному заместителю и в спиртах выступает как электроноакцептор. Спирты имеют большое сходство с водой, но алкильный заместитель увеличивает ковалентность связи ОН: R О-Н.
 -I +C В молекуле фенола –ОН-группа непосредственно связана с бензольным кольцом: фенол спирт
-I +C В молекуле фенола –ОН-группа непосредственно связана с бензольным кольцом: фенол спирт
 Аналогично молекулам спиртов -ОН группа в фенолах проявляет – I-эффект, являясь -электроноакцептором. Однако электронная пара, находящаяся на pz-орбитали, может вступать в сопряжение с -электронной системой бензола, отдавая электронную плотность в ядро и проявляя тем самым значительный +С-эффект. Поскольку -I +С , то –ОНгруппа в фенолах является сильным электронодонором (орто- и пара-ориентант в реакциях электрофильного замещения). Физические свойства Одноатомные спирты, содержащие до 16 атомов углерода – жидкости, более 16 – твердые вещества. Спирты являются высокоассоциированными жидкостями. Способность гидроксисоединений к образованию сетки водородных связей является следствием полярности связи О–Н и наличия неподеленных пар электронов на атоме кислорода:
Аналогично молекулам спиртов -ОН группа в фенолах проявляет – I-эффект, являясь -электроноакцептором. Однако электронная пара, находящаяся на pz-орбитали, может вступать в сопряжение с -электронной системой бензола, отдавая электронную плотность в ядро и проявляя тем самым значительный +С-эффект. Поскольку -I +С , то –ОНгруппа в фенолах является сильным электронодонором (орто- и пара-ориентант в реакциях электрофильного замещения). Физические свойства Одноатомные спирты, содержащие до 16 атомов углерода – жидкости, более 16 – твердые вещества. Спирты являются высокоассоциированными жидкостями. Способность гидроксисоединений к образованию сетки водородных связей является следствием полярности связи О–Н и наличия неподеленных пар электронов на атоме кислорода:
 Энергия простой водородной связи в спиртах 25 -26 к. Дж/моль. Ассоциаты могут быть линейными либо циклическими. n R-OH (ROH)n, где n – степень ассоциации, n = 2 30 и более. Ассоциация проявляется в высоких температурах кипения спиртов по сравнению с температурами кипения соответствующих углеводородов. Например, С 3 Н 8 – пропан, М=44, tкип=-42 о. С; С 2 Н 5 ОН – этанол, М=46, tкип=78 о. С. Чем менее разветвленным является заместитель, тем больше способность спирта образовывать ассоциаты и выше его температура кипения: первичный спирт вторичный спирт третичный спирт уменьшение температур кипения Температуры кипения изменяются в ряду: н-бутанол (117, 9 о. С) втор-бутанол (100 о. С) трет-бутанол (83 о. С).
Энергия простой водородной связи в спиртах 25 -26 к. Дж/моль. Ассоциаты могут быть линейными либо циклическими. n R-OH (ROH)n, где n – степень ассоциации, n = 2 30 и более. Ассоциация проявляется в высоких температурах кипения спиртов по сравнению с температурами кипения соответствующих углеводородов. Например, С 3 Н 8 – пропан, М=44, tкип=-42 о. С; С 2 Н 5 ОН – этанол, М=46, tкип=78 о. С. Чем менее разветвленным является заместитель, тем больше способность спирта образовывать ассоциаты и выше его температура кипения: первичный спирт вторичный спирт третичный спирт уменьшение температур кипения Температуры кипения изменяются в ряду: н-бутанол (117, 9 о. С) втор-бутанол (100 о. С) трет-бутанол (83 о. С).
 Многоатомные спирты еще более ассоциированы, поэтому при переходе от одноатомных к многоатомным спиртам температуры кипения и плавления резко возрастают (пропанол (97 о. С) < пропиленгликоль (189 о. С) < глицерин (290 о. С) ). Молекулы низших спиртов отличаются высокой полярностью ( ≈ 1, 7). Дипольный момент фенолов несколько ниже ( ≈ 1, 53 у С 6 Н 5 ОН). Низшие члены ряда спиртов хорошо растворимы в воде за счет образования водородных связей с молекулами воды:
Многоатомные спирты еще более ассоциированы, поэтому при переходе от одноатомных к многоатомным спиртам температуры кипения и плавления резко возрастают (пропанол (97 о. С) < пропиленгликоль (189 о. С) < глицерин (290 о. С) ). Молекулы низших спиртов отличаются высокой полярностью ( ≈ 1, 7). Дипольный момент фенолов несколько ниже ( ≈ 1, 53 у С 6 Н 5 ОН). Низшие члены ряда спиртов хорошо растворимы в воде за счет образования водородных связей с молекулами воды:
 Способность растворяться в воде уменьшается при переходе от многоатомных гидроксисоединений к одноатомным, а также с увеличением длины алкильной цепочки. Метанол, этанол, пропанол, изопропанол, этиленгликоль и глицерин смешиваются с водой в любых соотношениях. н-Бутанол растворим в воде при 25 о. С всего на 8%. Одноатомные спирты являются хорошими растворителями. Фенол – твердое белое кристаллическое вещество с ограниченной растворимостью в воде. Многоатомные фенолы хорошо растворимы в воде (размер полярной части молекулы увеличивается).
Способность растворяться в воде уменьшается при переходе от многоатомных гидроксисоединений к одноатомным, а также с увеличением длины алкильной цепочки. Метанол, этанол, пропанол, изопропанол, этиленгликоль и глицерин смешиваются с водой в любых соотношениях. н-Бутанол растворим в воде при 25 о. С всего на 8%. Одноатомные спирты являются хорошими растворителями. Фенол – твердое белое кристаллическое вещество с ограниченной растворимостью в воде. Многоатомные фенолы хорошо растворимы в воде (размер полярной части молекулы увеличивается).
 Методы получения одноатомных спиртов: 1. Спиртовое брожение глюкозы с образованием этилового спирта: 2 С 2 Н 5 ОН + 2 СО 2 С 6 Н 12 О 6 2. Гидролиз галогеналканов: С 2 Н 5 Cl + Н 2 О/Na. OH С 2 Н 5 ОН + Na. Cl 3. Гидратация алкенов: СН 2=СН-СН 3 + Н 2 О Этот способ применяют в промышленности для получения этанола и изопропанола. В случае других спиртов его применение в органическом синтезе нецелесообразно из-за большого числа побочных продуктов.
Методы получения одноатомных спиртов: 1. Спиртовое брожение глюкозы с образованием этилового спирта: 2 С 2 Н 5 ОН + 2 СО 2 С 6 Н 12 О 6 2. Гидролиз галогеналканов: С 2 Н 5 Cl + Н 2 О/Na. OH С 2 Н 5 ОН + Na. Cl 3. Гидратация алкенов: СН 2=СН-СН 3 + Н 2 О Этот способ применяют в промышленности для получения этанола и изопропанола. В случае других спиртов его применение в органическом синтезе нецелесообразно из-за большого числа побочных продуктов.
 4. Оксимеркурирование – демеркурирование алкенов: Направление протекания реакции соответствует правилу Марковникова, образуются вторичные или третичные спирты. 5. Гидроборирование алкенов с последующим окислением боранов: Таким способом получают первичные спирты против правила Марковникова.
4. Оксимеркурирование – демеркурирование алкенов: Направление протекания реакции соответствует правилу Марковникова, образуются вторичные или третичные спирты. 5. Гидроборирование алкенов с последующим окислением боранов: Таким способом получают первичные спирты против правила Марковникова.
 6. Синтезы с использованием реактивов Гриньяра: Эта реакция очень хорошо известна как метод синтеза спиртов. Она обратима, особенно в случае пространственно затрудненных субстратов. Из формальдегида образуются первичные спирты, из других альдегидов - вторичные спирты, а из кетонов третичные спирты.
6. Синтезы с использованием реактивов Гриньяра: Эта реакция очень хорошо известна как метод синтеза спиртов. Она обратима, особенно в случае пространственно затрудненных субстратов. Из формальдегида образуются первичные спирты, из других альдегидов - вторичные спирты, а из кетонов третичные спирты.
 7. Восстановление соединений с С=О связью гидридами металлов (Li. Al. H 4, Na. BH 4):
7. Восстановление соединений с С=О связью гидридами металлов (Li. Al. H 4, Na. BH 4):
 восстанавливают альдегиды до первичных") Оба реагента (Li. Al. H 4 и Na. BH 4) восстанавливают альдегиды до первичных спиртов, а кетоны до вторичных. Боргидрид натрия предпочтительнее вследствие меньшей стоимости и безопасности в обращении. Он может быть использован даже в водном и спиртовом растворах, тогда как алюмогидрид лития реагирует с водой и со спиртом со взрывом и, кроме того, разлагается со взрывом при нагревании выше 120°С в сухом состоянии: Алюмогидрид лития при 0 -10 o. С восстанавливает α, βненасыщенные альдегиды и кетоны до аллиловых спиртов, т. е. с сохранением углерод-углеродной двойной связи. Восстановлению в этом случае подвергается карбонильная группа:
Оба реагента (Li. Al. H 4 и Na. BH 4) восстанавливают альдегиды до первичных спиртов, а кетоны до вторичных. Боргидрид натрия предпочтительнее вследствие меньшей стоимости и безопасности в обращении. Он может быть использован даже в водном и спиртовом растворах, тогда как алюмогидрид лития реагирует с водой и со спиртом со взрывом и, кроме того, разлагается со взрывом при нагревании выше 120°С в сухом состоянии: Алюмогидрид лития при 0 -10 o. С восстанавливает α, βненасыщенные альдегиды и кетоны до аллиловых спиртов, т. е. с сохранением углерод-углеродной двойной связи. Восстановлению в этом случае подвергается карбонильная группа:
 8. ВОССТАНОВЛЕНИЕ СЛОЖНЫХ ЭФИРОВ И КАРБОНОВЫХ КИСЛОТ ДО ПЕРВИЧНЫХ СПИРТОВ Первичные спирты образуются при восстановлении сложных эфиров и карбоновых кислот алюмогидридом лития в эфире и ТГФ. Восстановление карбонильной группы комплексными гидридами относится к реакциям нуклеофильного присоединения, характерным для карбонильной группы. Роль нуклеофильного агента в этом случае выполняет гидрид-ион.
8. ВОССТАНОВЛЕНИЕ СЛОЖНЫХ ЭФИРОВ И КАРБОНОВЫХ КИСЛОТ ДО ПЕРВИЧНЫХ СПИРТОВ Первичные спирты образуются при восстановлении сложных эфиров и карбоновых кислот алюмогидридом лития в эфире и ТГФ. Восстановление карбонильной группы комплексными гидридами относится к реакциям нуклеофильного присоединения, характерным для карбонильной группы. Роль нуклеофильного агента в этом случае выполняет гидрид-ион.
 Химические свойства спиртов В молекулах спиртов и фенолов имеется 4 реакционных центра, по которым протекают: 1 – реакции с разрывом связи О-Н (взаимодействие с основаниями, образование сложных эфиров карбоновых кислот, ацилирование и алкилирование); 2 – реакции по неподеленной электронной паре (взаимодействие с кислотами, процессы ассоциации, комплексообразование); 3 – реакции по связи С-ОН (замещение ОН-группы на нуклеофил, межмолекулярная и внутримолекулярная дегидратация). Для фенолов такие реакции малохарактерны вследствие действия сильного +С-эффекта сопряжения –ОН группы с бензольным кольцом; 4 – реакции замещения водорода при -углеродном атоме, реакции окисления, дегидрирования (у фенолов отсутствуют); 5 – реакции с участием бензольного кольца (электрофильное замещение).
Химические свойства спиртов В молекулах спиртов и фенолов имеется 4 реакционных центра, по которым протекают: 1 – реакции с разрывом связи О-Н (взаимодействие с основаниями, образование сложных эфиров карбоновых кислот, ацилирование и алкилирование); 2 – реакции по неподеленной электронной паре (взаимодействие с кислотами, процессы ассоциации, комплексообразование); 3 – реакции по связи С-ОН (замещение ОН-группы на нуклеофил, межмолекулярная и внутримолекулярная дегидратация). Для фенолов такие реакции малохарактерны вследствие действия сильного +С-эффекта сопряжения –ОН группы с бензольным кольцом; 4 – реакции замещения водорода при -углеродном атоме, реакции окисления, дегидрирования (у фенолов отсутствуют); 5 – реакции с участием бензольного кольца (электрофильное замещение).
 I. Реакции с разрывом связи О-Н 1. Ионизация спиртов. RОН + ОН RО- + Н 2 О алкоголят- ион (алкоксид-ион) (слабее Н 2 О, кроме метанола) Спирты как слабые ОН-кислоты реагируют с щелочными, щелочноземельными металлами, алюминием, галлием и таллием с образованием алкоголятов: СН 3 ОН + Na СН 3 ОNa + ½ Н 2 метилат Na Алкоголяты RO- – очень сильные основания, легко гидролизуются с образованием спиртов. Кислотность спиртов (р. Ка) в водном растворе уменьшается в ряду: H 2 O (15, 7) СН 3 ОН (15, 5) первичный (С 2 Н 5 ОН, 15, 9) вторичный ((СН 3)2 СН-ОН, 16, 9) третичный ((СН 3)3 С-ОН, 19, 2).
I. Реакции с разрывом связи О-Н 1. Ионизация спиртов. RОН + ОН RО- + Н 2 О алкоголят- ион (алкоксид-ион) (слабее Н 2 О, кроме метанола) Спирты как слабые ОН-кислоты реагируют с щелочными, щелочноземельными металлами, алюминием, галлием и таллием с образованием алкоголятов: СН 3 ОН + Na СН 3 ОNa + ½ Н 2 метилат Na Алкоголяты RO- – очень сильные основания, легко гидролизуются с образованием спиртов. Кислотность спиртов (р. Ка) в водном растворе уменьшается в ряду: H 2 O (15, 7) СН 3 ОН (15, 5) первичный (С 2 Н 5 ОН, 15, 9) вторичный ((СН 3)2 СН-ОН, 16, 9) третичный ((СН 3)3 С-ОН, 19, 2).
 В газовой фазе наблюдается противоположная последовательность. При введении электроноакцепторных заместителей - фтора, хлора, цианогруппы и др. - величина р. Кa уменьшается, что соответствует усилению кислотных свойств: СН 3 СН 2 ОН р. Ка = 15, 9 CF 3 CH 2 OH р. Ка = 12, 4 2. Образование простых эфиров В мягких условиях при нагревании простейших первичных спиртов с 96%-й серной кислотой при 130 - 140 °С преимущественно получаются простые эфиры. Механизм этого превращения заключается в алкилировании первичного спирта либо под действием полуэфира серной кислоты, либо при взаимодействии с катионом алкоксония, кинетически оба этих механизма SN 2 -замещения неразличимы:
В газовой фазе наблюдается противоположная последовательность. При введении электроноакцепторных заместителей - фтора, хлора, цианогруппы и др. - величина р. Кa уменьшается, что соответствует усилению кислотных свойств: СН 3 СН 2 ОН р. Ка = 15, 9 CF 3 CH 2 OH р. Ка = 12, 4 2. Образование простых эфиров В мягких условиях при нагревании простейших первичных спиртов с 96%-й серной кислотой при 130 - 140 °С преимущественно получаются простые эфиры. Механизм этого превращения заключается в алкилировании первичного спирта либо под действием полуэфира серной кислоты, либо при взаимодействии с катионом алкоксония, кинетически оба этих механизма SN 2 -замещения неразличимы:
 Этим способом получают простейшие простые эфиры - диэтиловый, дипропиловый и дибутиловый эфиры и циклические простые эфиры, например тетрагидрофуран или диоксан. Вторичные и третичные спирты в этих условиях дегидратируются с образованием алкенов. Этот способ непригоден для получения несимметричных эфиров.
Этим способом получают простейшие простые эфиры - диэтиловый, дипропиловый и дибутиловый эфиры и циклические простые эфиры, например тетрагидрофуран или диоксан. Вторичные и третичные спирты в этих условиях дегидратируются с образованием алкенов. Этот способ непригоден для получения несимметричных эфиров.
: II. Реакции по неподеленной электронной паре атома кислорода.") 3. Образование сложных эфиров (реакция этерификации): II. Реакции по неподеленной электронной паре атома кислорода. 1. Протонирование спиртов сильными кислотами (HCl, H 2 SO 4 и др. ) R-OH + H+X- [R-OH 2+]Xалкилгидроксоний – ион С галогенидами и оксигалогенидами фосфора, серы, выступающими в качестве кислот Льюиса, спирты образуют донорно-акцепторные комплексы, которые в некоторых условиях могут подвергаться дальнейшим превращениям.
3. Образование сложных эфиров (реакция этерификации): II. Реакции по неподеленной электронной паре атома кислорода. 1. Протонирование спиртов сильными кислотами (HCl, H 2 SO 4 и др. ) R-OH + H+X- [R-OH 2+]Xалкилгидроксоний – ион С галогенидами и оксигалогенидами фосфора, серы, выступающими в качестве кислот Льюиса, спирты образуют донорно-акцепторные комплексы, которые в некоторых условиях могут подвергаться дальнейшим превращениям.
 2. Реакции комплексообразования, в которых молекулы спирта могут выступать в качестве простых лигандов, например [Cu(OAc)2(C 2 H 5 OH)n-2], [Zn(C 2 H 5 OH)6]2+(NO 3 -)2 III. Реакции с разрывом связи С-ОН 1. Нуклеофильное замещение ОН-группы на галоген Первичные спирты реагируют по механизму SN 2: СН 3 СН 2 ОН + HBr СН 3 СН 2 ОН 2+ + Br- СН 3 СН 2 Br + Н 2 О
2. Реакции комплексообразования, в которых молекулы спирта могут выступать в качестве простых лигандов, например [Cu(OAc)2(C 2 H 5 OH)n-2], [Zn(C 2 H 5 OH)6]2+(NO 3 -)2 III. Реакции с разрывом связи С-ОН 1. Нуклеофильное замещение ОН-группы на галоген Первичные спирты реагируют по механизму SN 2: СН 3 СН 2 ОН + HBr СН 3 СН 2 ОН 2+ + Br- СН 3 СН 2 Br + Н 2 О
 Третичные спирты реагируют по механизму SN 1: Сама по себе –ОН-группа является «плохой уходящей группой» , т. к. легче отщепляются группы – анионы сильных кислот. Группе –ОН соответствует молекула Н 2 О (слабая кислота), тогда как протонированной группе –ОН 2+ (хорошая уходящая группа) соответствует ион гидроксония Н 3 О+ (сильная кислота). Поэтому протонирование молекулы спирта приводит к увеличению скорости как SN 1, так и SN 2 -реакций. Реакционная способность галогенводородов уменьшается в ряду: HJ HBr HCl HF
Третичные спирты реагируют по механизму SN 1: Сама по себе –ОН-группа является «плохой уходящей группой» , т. к. легче отщепляются группы – анионы сильных кислот. Группе –ОН соответствует молекула Н 2 О (слабая кислота), тогда как протонированной группе –ОН 2+ (хорошая уходящая группа) соответствует ион гидроксония Н 3 О+ (сильная кислота). Поэтому протонирование молекулы спирта приводит к увеличению скорости как SN 1, так и SN 2 -реакций. Реакционная способность галогенводородов уменьшается в ряду: HJ HBr HCl HF
 Скорость реакции замещения резко снижается в ряду третичный > вторичный > первичный спирт. Для получения третичных алкилгалогенидов обычно достаточно насытить третичный спирт газообразным галогеноводородом при 0 -10 °С или обработать водной соляной, бромистоводородной или йодистоводородной кислотой в течение короткого промежутка времени при 0 -20 o. С. Для получения первичных и вторичных алкилбромидов и алкилйодидов обычно требуется нагревание смеси спирта, концентрированной бромистоводородной или йодистоводородной кислоты и концентрированной серной кислоты в течение нескольких часов.
Скорость реакции замещения резко снижается в ряду третичный > вторичный > первичный спирт. Для получения третичных алкилгалогенидов обычно достаточно насытить третичный спирт газообразным галогеноводородом при 0 -10 °С или обработать водной соляной, бромистоводородной или йодистоводородной кислотой в течение короткого промежутка времени при 0 -20 o. С. Для получения первичных и вторичных алкилбромидов и алкилйодидов обычно требуется нагревание смеси спирта, концентрированной бромистоводородной или йодистоводородной кислоты и концентрированной серной кислоты в течение нескольких часов.
 Хлорид-ион в гидроксилсодержащих растворителях сильно сольватирован и проявляет свойства более слабого нуклеофильного агента по сравнению с бромид- и йодид-ионами. Поэтому для получения алкилхлоридов при взаимодействии первичных спиртов с соляной кислотой используют электрофильный катализатор - безводный хлорид цинка. Смесь соляной кислоты и хлорида цинка носит название реактива Лукаса: RCH 2 OH + Zn. Cl 2 RCH 2 Cl + Zn(OH)Cl 2 -
Хлорид-ион в гидроксилсодержащих растворителях сильно сольватирован и проявляет свойства более слабого нуклеофильного агента по сравнению с бромид- и йодид-ионами. Поэтому для получения алкилхлоридов при взаимодействии первичных спиртов с соляной кислотой используют электрофильный катализатор - безводный хлорид цинка. Смесь соляной кислоты и хлорида цинка носит название реактива Лукаса: RCH 2 OH + Zn. Cl 2 RCH 2 Cl + Zn(OH)Cl 2 -
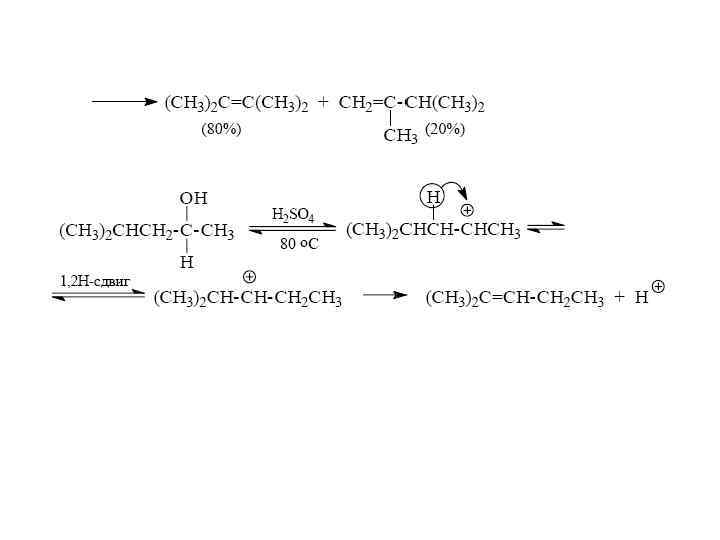
 Другой важной характерной особенностью процессов с участием карбокатионов являются перегруппировки. Атом водорода при соседнем атоме углерода может мигрировать с парой электронов в виде гидрид-иона к карбокатионному центру. Это смещение носит название 1, 2 -гидридного сдвига. Подобного рода перегруппировки наблюдаются очень часто, их роль особенно возрастает тогда, когда в результате 1, 2 -гидридного сдвига образуется более стабильный карбокатион:
Другой важной характерной особенностью процессов с участием карбокатионов являются перегруппировки. Атом водорода при соседнем атоме углерода может мигрировать с парой электронов в виде гидрид-иона к карбокатионному центру. Это смещение носит название 1, 2 -гидридного сдвига. Подобного рода перегруппировки наблюдаются очень часто, их роль особенно возрастает тогда, когда в результате 1, 2 -гидридного сдвига образуется более стабильный карбокатион:
 Например, при нагревании З-метилбутанола-2, насыщенного газообразным бромистым водородом, в качестве единственного продукта реакции образуется 2 -бром-2 -метилбутан вместо 2 бром-З-метилбутана - «нормального» продукта замещения гидроксила на галоген:
Например, при нагревании З-метилбутанола-2, насыщенного газообразным бромистым водородом, в качестве единственного продукта реакции образуется 2 -бром-2 -метилбутан вместо 2 бром-З-метилбутана - «нормального» продукта замещения гидроксила на галоген:
 Перегруппировка может происходить не только при 1, 2 миграции гидрид-иона, но также в результате скелетной изомеризации, когда мигрирует алкильная группа. Если структурные факторы способствуют образованию третичного карбокатиона, первичный спирт претерпевает перегруппировку Вагнера-Меервейна:
Перегруппировка может происходить не только при 1, 2 миграции гидрид-иона, но также в результате скелетной изомеризации, когда мигрирует алкильная группа. Если структурные факторы способствуют образованию третичного карбокатиона, первичный спирт претерпевает перегруппировку Вагнера-Меервейна:
 Пентанол-1 в тех же условиях образует только 1 -бромпентан без продуктов перегруппировки, и это превращение, несомненно, протекает по SN 2 -механизму. Однако первичные спирты с разветвленной цепью дают уже некоторое количество изомерного галогенида: Таким образом, замещение гидроксильной группы спиртов на галоген под действием галогеноводородов без изомеризации осуществляется только для третичных спиртов и н-алканолов-1.
Пентанол-1 в тех же условиях образует только 1 -бромпентан без продуктов перегруппировки, и это превращение, несомненно, протекает по SN 2 -механизму. Однако первичные спирты с разветвленной цепью дают уже некоторое количество изомерного галогенида: Таким образом, замещение гидроксильной группы спиртов на галоген под действием галогеноводородов без изомеризации осуществляется только для третичных спиртов и н-алканолов-1.
 Более эффективно замещение ОН-группы на галоген протекает при действии галогенидов фосфора: 3 ROH + PBr 3 3 R-Br + H 3 PO 3 Для превращения спиртов в алкилгалогениды применяют различные три- и пентагалогениды фосфора: РВr 3; РСl 5; РОС 13 или РI 3, получаемый из красного фосфора и йода непосредственно во время реакции. Первая стадия этого процесса по существу представляет собой нуклеофильное замещение у атома фосфора под действием спирта как нуклеофильного агента:
Более эффективно замещение ОН-группы на галоген протекает при действии галогенидов фосфора: 3 ROH + PBr 3 3 R-Br + H 3 PO 3 Для превращения спиртов в алкилгалогениды применяют различные три- и пентагалогениды фосфора: РВr 3; РСl 5; РОС 13 или РI 3, получаемый из красного фосфора и йода непосредственно во время реакции. Первая стадия этого процесса по существу представляет собой нуклеофильное замещение у атома фосфора под действием спирта как нуклеофильного агента:
 Замещение гидроксила на галоген под действием РВr 3, РI 3, РСl 5, РОСl 3 часто сопровождается изомеризацией и перегруппировками. Пентанол-3 в реакции с трехбромистым фосфором в эфире образует 85% 3 -бромпентана и 15% 2 бромпентана - продукта перегруппировки. Степень изомеризации и перегруппировки здесь ниже, чем в реакциях замещения под действием НВr и других галогеноводородов, но все-таки весьма велика: Тионилхлорид превращает первичные и вторичные спирты в алкилхлориды с выходом 70 -90%.
Замещение гидроксила на галоген под действием РВr 3, РI 3, РСl 5, РОСl 3 часто сопровождается изомеризацией и перегруппировками. Пентанол-3 в реакции с трехбромистым фосфором в эфире образует 85% 3 -бромпентана и 15% 2 бромпентана - продукта перегруппировки. Степень изомеризации и перегруппировки здесь ниже, чем в реакциях замещения под действием НВr и других галогеноводородов, но все-таки весьма велика: Тионилхлорид превращает первичные и вторичные спирты в алкилхлориды с выходом 70 -90%.
 2. Взаимодействие с кислородсодержащими неорганическими кислотами СН 3 СН 2 ОН + H 2 SO 4 [СН 3 СН 2 ОН 2+] HSO 4 - СН 3 СН 2 ОSO 3 Н этилсерная кислота (этилсульфат) 3. Дегидратация спиртов происходит при нагревании в концентрированной серной кислоте, фосфорной кислоте или в суперкислой среде - смеси пятифтористой сурьмы и фторсульфоновой кислоты. Главное условие – отсутствие в системе нуклеофилов.
2. Взаимодействие с кислородсодержащими неорганическими кислотами СН 3 СН 2 ОН + H 2 SO 4 [СН 3 СН 2 ОН 2+] HSO 4 - СН 3 СН 2 ОSO 3 Н этилсерная кислота (этилсульфат) 3. Дегидратация спиртов происходит при нагревании в концентрированной серной кислоте, фосфорной кислоте или в суперкислой среде - смеси пятифтористой сурьмы и фторсульфоновой кислоты. Главное условие – отсутствие в системе нуклеофилов.
 Реакционная способность спиртов уменьшается в ряду: третичный спирт вторичный спирт первичный спирт Дегидратацию третичных спиртов можно проводить уже в 20 -50% серной кислоте при 85 -100 о. С, первичные спирты подвергаются дегидратации в значительно более жестких условиях (96% H 2 SO 4 при 170 -190 о. С). Дегидратация спиртов протекает также в присутствии такого водоотнимающего средства, как Al 2 O 3: СН 3 СН 2 ОН СН 2=СН 2
Реакционная способность спиртов уменьшается в ряду: третичный спирт вторичный спирт первичный спирт Дегидратацию третичных спиртов можно проводить уже в 20 -50% серной кислоте при 85 -100 о. С, первичные спирты подвергаются дегидратации в значительно более жестких условиях (96% H 2 SO 4 при 170 -190 о. С). Дегидратация спиртов протекает также в присутствии такого водоотнимающего средства, как Al 2 O 3: СН 3 СН 2 ОН СН 2=СН 2
 Дегидратация многих спиртов сопровождается перегруппировкой, заключающейся в 1, 2 -миграции алкильной группы или гидрид-иона. Такие перегруппировки типичны для процессов с участием карбокатионов в качестве промежуточных частиц. Наблюдаемый порядок уменьшения реакционной способности спиртов: третичный > вторичный > первичный и наличие перегруппировок согласуются с карбокатионным Е 1 механизмом дегидратации:
Дегидратация многих спиртов сопровождается перегруппировкой, заключающейся в 1, 2 -миграции алкильной группы или гидрид-иона. Такие перегруппировки типичны для процессов с участием карбокатионов в качестве промежуточных частиц. Наблюдаемый порядок уменьшения реакционной способности спиртов: третичный > вторичный > первичный и наличие перегруппировок согласуются с карбокатионным Е 1 механизмом дегидратации:
 В более мягких условиях при нагревании простейших первичных спиртов с 96%-й серной кислотой при 130 -140 °С преимущественно получаются простые эфиры: Вторичные и третичные спирты в этих условиях дегидратируются с образованием алкенов.
В более мягких условиях при нагревании простейших первичных спиртов с 96%-й серной кислотой при 130 -140 °С преимущественно получаются простые эфиры: Вторичные и третичные спирты в этих условиях дегидратируются с образованием алкенов.
 окисление хлором б) первичные спирты") IV. Реакции по -углеродному атому 1. Окисление спиртов: а) окисление хлором б) первичные спирты окисляются различными окислителями до альдегидов или карбоновых кислот:
IV. Реакции по -углеродному атому 1. Окисление спиртов: а) окисление хлором б) первичные спирты окисляются различными окислителями до альдегидов или карбоновых кислот:
 Третичные спирты в обычных условиях не окисляются, а в очень жестких условиях их окисление сопровождается деструкцией углеродного скелета.
Третичные спирты в обычных условиях не окисляются, а в очень жестких условиях их окисление сопровождается деструкцией углеродного скелета.
 Чаще всего, для окисления вторичных спиртов до кетонов в качестве окислителя используют реагент Джонса - раствор строго рассчитанного количества Сr. О 3 в водной серной кислоте. Вторичные спирты, содержащие при этом двойную или тройную связь, быстро окисляются до кетонов без затрагивания кратных связей: Первичные спирты окисляются реагентом Джонса до карбоновых кислот.
Чаще всего, для окисления вторичных спиртов до кетонов в качестве окислителя используют реагент Джонса - раствор строго рассчитанного количества Сr. О 3 в водной серной кислоте. Вторичные спирты, содержащие при этом двойную или тройную связь, быстро окисляются до кетонов без затрагивания кратных связей: Первичные спирты окисляются реагентом Джонса до карбоновых кислот.
 Дальнейшее окисление кетонов возможно с разрывом углеродных связей и образованием смеси карбоновых кислот: (в общем случае 4 молекулы карбоновых кислот) Третичные спирты в щелочной среде не окисляются, в кислой среде быстро отщепляют воду с образованием алкенов, которые затем подвергаются окислению по двойной связи. 2. Дегидрирование спиртов
Дальнейшее окисление кетонов возможно с разрывом углеродных связей и образованием смеси карбоновых кислот: (в общем случае 4 молекулы карбоновых кислот) Третичные спирты в щелочной среде не окисляются, в кислой среде быстро отщепляют воду с образованием алкенов, которые затем подвергаются окислению по двойной связи. 2. Дегидрирование спиртов
 ДВУХАТОМНЫЕ СПИРТЫ Получение: 1. Наиболее известными способами получения 1, 2 -диолов являются разнообразные реакции стереоселективного син- или анти-гидроксилирования алкенов. 2. Классическим методом получения симметричных 1, 2 диолов является восстановительная димеризация кетонов:
ДВУХАТОМНЫЕ СПИРТЫ Получение: 1. Наиболее известными способами получения 1, 2 -диолов являются разнообразные реакции стереоселективного син- или анти-гидроксилирования алкенов. 2. Классическим методом получения симметричных 1, 2 диолов является восстановительная димеризация кетонов:
 Поскольку диол, получаемый этим методом из ацетона, имеет тривиальное название пинакон, эта реакция получила название пинаконовое восстановление: 3. Окисление алкенов
Поскольку диол, получаемый этим методом из ацетона, имеет тривиальное название пинакон, эта реакция получила название пинаконовое восстановление: 3. Окисление алкенов
 СВОЙСТВА ДИОЛОВ 1. Обладают более сильными кислотными свойствами по сравнению с одноатомными спиртами, реагируют с Na. ОН: НО-СН 2 -ОН + 2 Na. ОН Na. О-СН 2 -ОNa + 2 Н 2 О 2. Образование хелатных комплексов 2 НО-СН 2 -ОН гликолят меди, темно-синего цвета - качественная реакция на многоатомные спирты
СВОЙСТВА ДИОЛОВ 1. Обладают более сильными кислотными свойствами по сравнению с одноатомными спиртами, реагируют с Na. ОН: НО-СН 2 -ОН + 2 Na. ОН Na. О-СН 2 -ОNa + 2 Н 2 О 2. Образование хелатных комплексов 2 НО-СН 2 -ОН гликолят меди, темно-синего цвета - качественная реакция на многоатомные спирты
 2. Образование сложных эфиров 2 НО-СН 2 -ОН + 2 HNO 3 + 2 Н 2 О динитрат этиленгликоля
2. Образование сложных эфиров 2 НО-СН 2 -ОН + 2 HNO 3 + 2 Н 2 О динитрат этиленгликоля
 3. Реакции дегидратации При дегидратации диолов возможно протекание различных реакций, которые часто приводят к образованию сложной смеси продуктов, состав которой зависит от структуры диола, природы дегидратирующего агента и условий реакции. Обсуждение будет сосредоточено главным образом на тех реакциях, механизм которых надежно установлен и которые играют заметную роль в органическом синтезе. Дегидратация 1, 2 диолов может осуществляться по трем принципиально различным направлениям: 1) дегидратация до диенов; 2) дегидратация, сопровождаемая перегруппировкой, - так называемая «пинаколиновая перегруппировка» ; 3) образование циклических эфиров и эпоксидов. Все эти реакции катализируются кислотными агентами, поэтому в общем случае все три направления конкурируют друг с другом.
3. Реакции дегидратации При дегидратации диолов возможно протекание различных реакций, которые часто приводят к образованию сложной смеси продуктов, состав которой зависит от структуры диола, природы дегидратирующего агента и условий реакции. Обсуждение будет сосредоточено главным образом на тех реакциях, механизм которых надежно установлен и которые играют заметную роль в органическом синтезе. Дегидратация 1, 2 диолов может осуществляться по трем принципиально различным направлениям: 1) дегидратация до диенов; 2) дегидратация, сопровождаемая перегруппировкой, - так называемая «пинаколиновая перегруппировка» ; 3) образование циклических эфиров и эпоксидов. Все эти реакции катализируются кислотными агентами, поэтому в общем случае все три направления конкурируют друг с другом.
 внутримолекулярная дегидратация: виниловый спирт,") Протекают при нагревании даже в присутствии слабых водоотнимающих средств 1) внутримолекулярная дегидратация: виниловый спирт, неустойчив акролеин (бесцветный газ, ядовит, оказывает раздражающее действие)
Протекают при нагревании даже в присутствии слабых водоотнимающих средств 1) внутримолекулярная дегидратация: виниловый спирт, неустойчив акролеин (бесцветный газ, ядовит, оказывает раздражающее действие)
 Дегидратация дитретичных, дивторичных и даже первично-третичных 1, 2 -диолов, катализируемая серной кислотой, n-толуолсульфокислотой,") 2) Дегидратация дитретичных, дивторичных и даже первично-третичных 1, 2 -диолов, катализируемая серной кислотой, n-толуолсульфокислотой, кислотами Льюиса (ВF 3 и др. ), сопровождается 1, 2 -миграцией алкильной, арильной группы или гидрид-иона. Продуктами перегруппировки являются кетоны или альдегиды. Эта перегруппировка была открыта Р. Фиттигом в 1859 г. при дегидратации пинакона в пинаколин с помощью концентрированной серной кислоты, поэтому она получила название пинаколиновой перегруппировки. При действии серной кислоты замещенные 1, 2 -диолы подвергаются дегидратации не с образованием соответствующих сопряженных алкадиенов (как это происходит в случае нагревания с HBr или на окиси алюминия при t 450 о. С), а с 1, 2 -миграцией алкильной, арильной группы или гидрид-иона и образованием альдегида или кетона:
2) Дегидратация дитретичных, дивторичных и даже первично-третичных 1, 2 -диолов, катализируемая серной кислотой, n-толуолсульфокислотой, кислотами Льюиса (ВF 3 и др. ), сопровождается 1, 2 -миграцией алкильной, арильной группы или гидрид-иона. Продуктами перегруппировки являются кетоны или альдегиды. Эта перегруппировка была открыта Р. Фиттигом в 1859 г. при дегидратации пинакона в пинаколин с помощью концентрированной серной кислоты, поэтому она получила название пинаколиновой перегруппировки. При действии серной кислоты замещенные 1, 2 -диолы подвергаются дегидратации не с образованием соответствующих сопряженных алкадиенов (как это происходит в случае нагревания с HBr или на окиси алюминия при t 450 о. С), а с 1, 2 -миграцией алкильной, арильной группы или гидрид-иона и образованием альдегида или кетона:
 Этиленгликоль и другие первично-вторичные 1, 2 -диолы общей формулы R-СН(ОН)-СН 2 ОН при") 3) Этиленгликоль и другие первично-вторичные 1, 2 -диолы общей формулы R-СН(ОН)-СН 2 ОН при нагреваниия с концентрированной серной или 85%-й фосфорной кислотой или п-толуолсульфокислотой дают 1, 4 -диоксаны - циклические простые эфиры с двумя атомами кислорода. Циклодегидратация 1, 4 -диолов и 1, 5 -диолов в тех же условиях служит наиболее важным способом получения производных тетрагидрофурана и тетрагидропирана соответственно:
3) Этиленгликоль и другие первично-вторичные 1, 2 -диолы общей формулы R-СН(ОН)-СН 2 ОН при нагреваниия с концентрированной серной или 85%-й фосфорной кислотой или п-толуолсульфокислотой дают 1, 4 -диоксаны - циклические простые эфиры с двумя атомами кислорода. Циклодегидратация 1, 4 -диолов и 1, 5 -диолов в тех же условиях служит наиболее важным способом получения производных тетрагидрофурана и тетрагидропирана соответственно:
 4. Окисление
4. Окисление
 ФЕНОЛЫ Фенолами называются соединения, у которых гидроксильная группа присоединена непосредственно к ароматическому кольцу бензола.
ФЕНОЛЫ Фенолами называются соединения, у которых гидроксильная группа присоединена непосредственно к ароматическому кольцу бензола.
 Соединения, содержащие гидроксильную группу у конденсированных ароматических соединений, называют нафтолами, фенантролами, антролами и т. д. , например:
Соединения, содержащие гидроксильную группу у конденсированных ароматических соединений, называют нафтолами, фенантролами, антролами и т. д. , например:
 Получение фенолов: 1. Щелочное плавление солей ароматических сульфокислот Точный механизм реакции в расплаве двух ионных соединений неизвестен, ее следует отнести к процессам нуклеофильного ароматического замещения, где гидроксид-ион играет роль нуклеофильного агента, а сульфит-ион - уходящей группы:
Получение фенолов: 1. Щелочное плавление солей ароматических сульфокислот Точный механизм реакции в расплаве двух ионных соединений неизвестен, ее следует отнести к процессам нуклеофильного ароматического замещения, где гидроксид-ион играет роль нуклеофильного агента, а сульфит-ион - уходящей группы:
 Для получения самого фенола метод щелочного плавления в настоящее время не используется, но он широко используется для получения 2 -нафтола , резорцина, ализарина и других фенолов. Метод щелочного плавления часто употребляется для замещения одной сульфогруппы на гидроксил в ди- и трисульфокислотах нафтолов и аминонафтолов. 2. Гидролиз галогенопроизводных аренов Арилгалогениды, не содержащие активирующих электроноакцепторных группировок, вступают в реакцию обмена в очень жестких условиях. Однако при нагревании хлорбензола с 15 -20%-ным водным раствором гидроксида натрия при 360 -390 о. С и давлении 28 м. Па (280 -300 атм) образуется фенол.
Для получения самого фенола метод щелочного плавления в настоящее время не используется, но он широко используется для получения 2 -нафтола , резорцина, ализарина и других фенолов. Метод щелочного плавления часто употребляется для замещения одной сульфогруппы на гидроксил в ди- и трисульфокислотах нафтолов и аминонафтолов. 2. Гидролиз галогенопроизводных аренов Арилгалогениды, не содержащие активирующих электроноакцепторных группировок, вступают в реакцию обмена в очень жестких условиях. Однако при нагревании хлорбензола с 15 -20%-ным водным раствором гидроксида натрия при 360 -390 о. С и давлении 28 м. Па (280 -300 атм) образуется фенол.
 позволяет проводить региоселективное замещение галогена на гидроксил без примеси какого-либо") Применение солей меди (II) позволяет проводить региоселективное замещение галогена на гидроксил без примеси какого-либо другого изомерного фенола. Введение в молекулу арилгалогенида электроноакцепторных заместителей в орто- или пара-положения увеличивают скорость обмена галогена на гидроксил в некатализируемой реакции.
Применение солей меди (II) позволяет проводить региоселективное замещение галогена на гидроксил без примеси какого-либо другого изомерного фенола. Введение в молекулу арилгалогенида электроноакцепторных заместителей в орто- или пара-положения увеличивают скорость обмена галогена на гидроксил в некатализируемой реакции.
 3. Гидролиз солей диазония Универсальным методом замены аминогруппы на гидроксил в ароматическом ряду является диазотирование первичного амина с последующими разложением соли диазония в водном растворе серной кислоты. Принято считать, что замещение диазогруппы на гидроксил протекает по SN 1 -механизму, крайне редко реализующемуся для других реакций в ароматическом ряду. Наилучшие результаты для получения оптимально высокого выхода фенолов достигается при постепенном введении раствора соли диазония в кипящий раствор серной кислоты.
3. Гидролиз солей диазония Универсальным методом замены аминогруппы на гидроксил в ароматическом ряду является диазотирование первичного амина с последующими разложением соли диазония в водном растворе серной кислоты. Принято считать, что замещение диазогруппы на гидроксил протекает по SN 1 -механизму, крайне редко реализующемуся для других реакций в ароматическом ряду. Наилучшие результаты для получения оптимально высокого выхода фенолов достигается при постепенном введении раствора соли диазония в кипящий раствор серной кислоты.
 4. Кумольный способ Современный промышленный метод получения фенола заключается в кислотно-катализируемом разложении гидропероксида кумола. Исходное вещество для всего цикла превращений - кумол получается в очень больших количествах при алкилировании бензола пропиленом по Фриделю-Крафтсу. Далее кумол окисляют кислородом воздуха при 100 -130 о. С до гидропероксида кумола.
4. Кумольный способ Современный промышленный метод получения фенола заключается в кислотно-катализируемом разложении гидропероксида кумола. Исходное вещество для всего цикла превращений - кумол получается в очень больших количествах при алкилировании бензола пропиленом по Фриделю-Крафтсу. Далее кумол окисляют кислородом воздуха при 100 -130 о. С до гидропероксида кумола.
 Третья, заключительная стадия всего процесса, по своему механизму напоминает перегруппировки карбкатионов:
Третья, заключительная стадия всего процесса, по своему механизму напоминает перегруппировки карбкатионов:
 Разложение гидропероксида кумола до фенола и ацетона проводят в присутствии 1% водной серной кислоты при 50 -90 о. С. Общая схема процесса: Применяется только для получения незамещенного фенола.
Разложение гидропероксида кумола до фенола и ацетона проводят в присутствии 1% водной серной кислоты при 50 -90 о. С. Общая схема процесса: Применяется только для получения незамещенного фенола.
 В целом это очень экономичный способ получения одновременно двух важнейших продуктов - ацетона и фенола. Аналогично в результате окисления пара- и метадиизопропилбензолов - побочных продуктов крупнотоннажного производства кумола, образуются бисгидропероксиды. При их разложении получаются соответственно гидрохинон и резорцин.
В целом это очень экономичный способ получения одновременно двух важнейших продуктов - ацетона и фенола. Аналогично в результате окисления пара- и метадиизопропилбензолов - побочных продуктов крупнотоннажного производства кумола, образуются бисгидропероксиды. При их разложении получаются соответственно гидрохинон и резорцин.
 Фенолы проявляют схожие со спиртами химические свойства. 1. Взаимодействие с основаниями протекает легче, чем у спиртов, т. е. фенолы являются более сильными кислотами, чем многоатомные и одноатомные спирты (р. Ка фенола в воде составляет 9, 8), образуют феноляты уже при действии таких оснований, как Na. OH или KOH. Фенолы на восемь и более порядков по кислотности превосходят спирты.
Фенолы проявляют схожие со спиртами химические свойства. 1. Взаимодействие с основаниями протекает легче, чем у спиртов, т. е. фенолы являются более сильными кислотами, чем многоатомные и одноатомные спирты (р. Ка фенола в воде составляет 9, 8), образуют феноляты уже при действии таких оснований, как Na. OH или KOH. Фенолы на восемь и более порядков по кислотности превосходят спирты.
 2. Образование фенолята железа – качественная реакция на фенолы: соль – фенолят железа, ярко-красного цвета 3. Ацилирование и алкилирование фенолов
2. Образование фенолята железа – качественная реакция на фенолы: соль – фенолят железа, ярко-красного цвета 3. Ацилирование и алкилирование фенолов
 Протекают не в бензольное кольцо, как SE-реакции, а по группе –ОН по нуклеофильному механизму. Нуклеофильные свойства проявляет атом кислорода молекулы фенола, центр атаки нуклеофила – sp 2 -гибридный атом углерода ангидрида или галогенангидрида кислоты (ацилирующего агента в реакции ацилирования), или атом углерода, связанный с атомом галогена (в реакции алкилирования). Ариловые эфиры карбоновых кислот нельзя получать прямой этерификацией фенолов карбоновыми кислотами. Обратимая реакция фенола с уксусной кислотой эндотермична в отличие от реакции этерификации спиртов, которая экзотермична.
Протекают не в бензольное кольцо, как SE-реакции, а по группе –ОН по нуклеофильному механизму. Нуклеофильные свойства проявляет атом кислорода молекулы фенола, центр атаки нуклеофила – sp 2 -гибридный атом углерода ангидрида или галогенангидрида кислоты (ацилирующего агента в реакции ацилирования), или атом углерода, связанный с атомом галогена (в реакции алкилирования). Ариловые эфиры карбоновых кислот нельзя получать прямой этерификацией фенолов карбоновыми кислотами. Обратимая реакция фенола с уксусной кислотой эндотермична в отличие от реакции этерификации спиртов, которая экзотермична.
 4. Реакции по неподеленной электронной паре, например протонирование, протекают сложнее, чем у спиртов. 5. Для фенолов малохарактерны реакции с разрывом связи С-О, т. к. сопряжение неподеленной электронной пары атома кислорода с pz-электронами бензольного ядра приводит к упрочнению этой связи. 6. Вследствие сильного электронодонорного влияния группы ОН на бензольное ядро реакции электрофильного замещения (SE 2) протекают очень легко, зачастую сразу в несколько положений:
4. Реакции по неподеленной электронной паре, например протонирование, протекают сложнее, чем у спиртов. 5. Для фенолов малохарактерны реакции с разрывом связи С-О, т. к. сопряжение неподеленной электронной пары атома кислорода с pz-электронами бензольного ядра приводит к упрочнению этой связи. 6. Вследствие сильного электронодонорного влияния группы ОН на бензольное ядро реакции электрофильного замещения (SE 2) протекают очень легко, зачастую сразу в несколько положений:
 Нитруют фенол по тем же причинам в очень мягких условиях (разбавленной азотной кислотой при охлаждении): 35% 15% Их легко разделить с помощью перегонки с водяным паром, где летучим оказывается только орто-изомер. Для введения в молекулу фенола только одного атома галогена используют галогенирование в неполярной среде:
Нитруют фенол по тем же причинам в очень мягких условиях (разбавленной азотной кислотой при охлаждении): 35% 15% Их легко разделить с помощью перегонки с водяным паром, где летучим оказывается только орто-изомер. Для введения в молекулу фенола только одного атома галогена используют галогенирование в неполярной среде:
 Моносульфирование фенола серной кислотой приводит к образованию смеси орто- и пара-изомеров гидроксибензолсульфоксилоты. При 20 о. С в реакционной смеси содержится 49% орто-изомера и 51% пара-изомера, тогда как при 120 о. С доля пара-изомера возрастает до 96%.
Моносульфирование фенола серной кислотой приводит к образованию смеси орто- и пара-изомеров гидроксибензолсульфоксилоты. При 20 о. С в реакционной смеси содержится 49% орто-изомера и 51% пара-изомера, тогда как при 120 о. С доля пара-изомера возрастает до 96%.
 Нитрозирование фенолов осуществляется с помощью азотистой кислоты в воде или в уксусной кислоте для тех фенолов, которые совершенно нерастворимы в воде. Нитрозирование фенолов отличается очень высокой региоселективностью в пара-положение по отношению к гидроксильной группе.
Нитрозирование фенолов осуществляется с помощью азотистой кислоты в воде или в уксусной кислоте для тех фенолов, которые совершенно нерастворимы в воде. Нитрозирование фенолов отличается очень высокой региоселективностью в пара-положение по отношению к гидроксильной группе.
 Алкилирование и ацилирование фенолов по Фриделю. Крафтсу Фенолы алкилируются в кольцо под действием самых разнообразных алкилирующих агентов: алкенов, спиртов и алкилгалогенидов в условиях кислотного катализа. Ацилирование фенолов в классических условиях реакции Фриделя-Крафтса комплексом ацилгалогенида и хлорида алюминия приводит к неудовлетворительным результатам, так как ацилированию подвергается гидроксильная группа фенола. Как одну из разновидностей реакции ацилирования по Фриделю-Крафтсу следует рассматривать конденсацию фенолов с фталевым ангидридом в присутствии серной кислоты или хлорида цинка (А. Байер, 1874 год). В этом случае две молекулы фенола конденсируются с одной молекулой фталевого ангидрида с образованием производных трифенилметана, называемых фталеинами.
Алкилирование и ацилирование фенолов по Фриделю. Крафтсу Фенолы алкилируются в кольцо под действием самых разнообразных алкилирующих агентов: алкенов, спиртов и алкилгалогенидов в условиях кислотного катализа. Ацилирование фенолов в классических условиях реакции Фриделя-Крафтса комплексом ацилгалогенида и хлорида алюминия приводит к неудовлетворительным результатам, так как ацилированию подвергается гидроксильная группа фенола. Как одну из разновидностей реакции ацилирования по Фриделю-Крафтсу следует рассматривать конденсацию фенолов с фталевым ангидридом в присутствии серной кислоты или хлорида цинка (А. Байер, 1874 год). В этом случае две молекулы фенола конденсируются с одной молекулой фталевого ангидрида с образованием производных трифенилметана, называемых фталеинами.
 При р. Н выше 9 водный раствор фенолфталеина окрашивается в малиновый цвет в результате расщепления лактонного цикла и образования дианиона.
При р. Н выше 9 водный раствор фенолфталеина окрашивается в малиновый цвет в результате расщепления лактонного цикла и образования дианиона.
 При конденсации фталевого ангидрида с резорцином образуется желто-зеленый флуоресцеин, широко используемый в качестве флуоресцирующего средства.
При конденсации фталевого ангидрида с резорцином образуется желто-зеленый флуоресцеин, широко используемый в качестве флуоресцирующего средства.
 Конденсация фенолов с альдегидами и кетонами Фенолы реагируют с формальдегидом в водном растворе в присутствии основания с образованием полимерного продукта, получившего название феноло-формальдегидной смолы, карболита или бакелита. В 1909 году Л. Бакелунд запатентовал способ получения этого первого синтетического высокомолекулярного соединения, которое сразу же нашло широкое применение в различных областях машиностроения, электротехники и быта, например, при изготовлении корпусов телефонов, электрических выключателей и т. д.
Конденсация фенолов с альдегидами и кетонами Фенолы реагируют с формальдегидом в водном растворе в присутствии основания с образованием полимерного продукта, получившего название феноло-формальдегидной смолы, карболита или бакелита. В 1909 году Л. Бакелунд запатентовал способ получения этого первого синтетического высокомолекулярного соединения, которое сразу же нашло широкое применение в различных областях машиностроения, электротехники и быта, например, при изготовлении корпусов телефонов, электрических выключателей и т. д.
 Карбоксилирование феноксид-ионов - реакция Кольбе Оригинальный метод введения карбоксильной группы в ароматическое кольцо был открыт Г. Кольбе в 1860 году. При нагревании сухих фенолятов натрия или лития с СО 2 при 150180 о. С и давлении 5 атм, образуются натриевые или литиевые соли салициловой кислоты. В аналогичных условиях из фенолятов калия, рубидия и цезия получаются только соли парагидроксибензойной кислоты.
Карбоксилирование феноксид-ионов - реакция Кольбе Оригинальный метод введения карбоксильной группы в ароматическое кольцо был открыт Г. Кольбе в 1860 году. При нагревании сухих фенолятов натрия или лития с СО 2 при 150180 о. С и давлении 5 атм, образуются натриевые или литиевые соли салициловой кислоты. В аналогичных условиях из фенолятов калия, рубидия и цезия получаются только соли парагидроксибензойной кислоты.
 7. Окисление фенолов. Окисление пространственно незатрудненных фенолов относится к числу сложных, многостадийных процессов, механизм которых мало изучен. Очевидно лишь то, что механизм окисления может сильно меняться в зависимости от природы одно- или двухэлектронного окислителя. Сам фенол при окислении двухэлектронным окислителем - бихроматом натрия или Mn. O 2 в серной кислоте образует с удовлетворительным выходом пара-хинон.
7. Окисление фенолов. Окисление пространственно незатрудненных фенолов относится к числу сложных, многостадийных процессов, механизм которых мало изучен. Очевидно лишь то, что механизм окисления может сильно меняться в зависимости от природы одно- или двухэлектронного окислителя. Сам фенол при окислении двухэлектронным окислителем - бихроматом натрия или Mn. O 2 в серной кислоте образует с удовлетворительным выходом пара-хинон.
 Резорцин окисляется очень медленно и в жестких условиях, пирокатехин и гидрохинон - легко. При окислении пирокатехина в качестве окислителя используется оксид серебра в эфире в присутствии сульфата натрия для связывания выделяющейся при окислении воды. Гидрохинон при окислении дает п-бензохинон. В качестве окислителей можно использовать самые разнообразные реагенты: дихромат натрия, ферроцианид калия, перкислоты, оксид серебра, тетраокись азота и ряд других окислителей.
Резорцин окисляется очень медленно и в жестких условиях, пирокатехин и гидрохинон - легко. При окислении пирокатехина в качестве окислителя используется оксид серебра в эфире в присутствии сульфата натрия для связывания выделяющейся при окислении воды. Гидрохинон при окислении дает п-бензохинон. В качестве окислителей можно использовать самые разнообразные реагенты: дихромат натрия, ферроцианид калия, перкислоты, оксид серебра, тетраокись азота и ряд других окислителей.
 п-бензохинон при взаимодействии с молекулами гидрохинона выпадает в осадок в виде темно-зеленых кристаллов хингидрона: гидрохинон 1, 4 -бензохинон хингидрон Хингидрон представляет собой классический пример молекулярных комплексов с переносом заряда, где один компонент служит донором, а другой является акцептором электрона.
п-бензохинон при взаимодействии с молекулами гидрохинона выпадает в осадок в виде темно-зеленых кристаллов хингидрона: гидрохинон 1, 4 -бензохинон хингидрон Хингидрон представляет собой классический пример молекулярных комплексов с переносом заряда, где один компонент служит донором, а другой является акцептором электрона.
 ХИНОНЫ Хиноны по своей структуре являются циклогексадиенонами, но их название происходит от ароматических углеводородов: бензохинон от бензола, толухинон от толуола, нафтохинон от нафталина и т. д. Цифры в начале названия обозначают положение двух карбонильных групп.
ХИНОНЫ Хиноны по своей структуре являются циклогексадиенонами, но их название происходит от ароматических углеводородов: бензохинон от бензола, толухинон от толуола, нафтохинон от нафталина и т. д. Цифры в начале названия обозначают положение двух карбонильных групп.
 Многие производные хинонов составляют важную группу природных веществ - красителей, пигментов, антибиотиков, витаминов и т. д.
Многие производные хинонов составляют важную группу природных веществ - красителей, пигментов, антибиотиков, витаминов и т. д.
 Получение хинонов Хиноны получают окислением одно- и двухатомных фенолов, аминов и диаминов ароматического ряда. Химические свойства хинонов 1. Наиболее важной реакцией хинонов является их двухстадийное восстановление до двухатомных фенолов.
Получение хинонов Хиноны получают окислением одно- и двухатомных фенолов, аминов и диаминов ароматического ряда. Химические свойства хинонов 1. Наиболее важной реакцией хинонов является их двухстадийное восстановление до двухатомных фенолов.
 2. Легкость восстановления хинонов до фенола открывает возможность для использования хинонов в качестве дегидрирующих агентов. Для этой цели выбирают хиноны с высоким окислительно-восстановительным потенциалом, такие как 2, 3, 5, 6 -тетрахлор-1, 4 -бензохинон (хлоранил); 2, 3 -дихлор-5, 6 дициано-1, 4 -бензохинон (ДДХ), дифенохинон. Дегидрированию подвергаются дигидроароматические соединения ряда бензола и тетрагидропроизводные ряда нафталина, антрацена, гетероциклических соединений, тропилиден и т. д.
2. Легкость восстановления хинонов до фенола открывает возможность для использования хинонов в качестве дегидрирующих агентов. Для этой цели выбирают хиноны с высоким окислительно-восстановительным потенциалом, такие как 2, 3, 5, 6 -тетрахлор-1, 4 -бензохинон (хлоранил); 2, 3 -дихлор-5, 6 дициано-1, 4 -бензохинон (ДДХ), дифенохинон. Дегидрированию подвергаются дигидроароматические соединения ряда бензола и тетрагидропроизводные ряда нафталина, антрацена, гетероциклических соединений, тропилиден и т. д.
 На протяжении длительного времени хиноны привлекали к себе интерес в производстве огромного количества высококачественных антрахиноновых красителей. Они широко использовались в качестве дегидрирующих агентов. В настоящее время интерес к этому классу соединений снова возрос после того, как было установлено, что целая группа хинонов играет жизненно важную роль переносчика электронов в дыхательных и фотохимических цепях биологических систем. В живых организмах эту роль транспорта электронов в дыхательных цепях в клетках выполняет группа коферментов Q, называемых убихинонами. В природе встречается несколько коферментов Q. Они отличаются друг от друга лишь числом изопреновых единиц, связанных с бензохиноновым кольцом. В организме человека важную роль играет кофермент Q 10.
На протяжении длительного времени хиноны привлекали к себе интерес в производстве огромного количества высококачественных антрахиноновых красителей. Они широко использовались в качестве дегидрирующих агентов. В настоящее время интерес к этому классу соединений снова возрос после того, как было установлено, что целая группа хинонов играет жизненно важную роль переносчика электронов в дыхательных и фотохимических цепях биологических систем. В живых организмах эту роль транспорта электронов в дыхательных цепях в клетках выполняет группа коферментов Q, называемых убихинонами. В природе встречается несколько коферментов Q. Они отличаются друг от друга лишь числом изопреновых единиц, связанных с бензохиноновым кольцом. В организме человека важную роль играет кофермент Q 10.
 ПРОСТЫЕ ЭФИРЫ Номенклатура и классификация I. Нециклические: диалкиловые эфиры R-O-R (напр. , CH 3 -O-CH 3 диметиловый эфир, метоксиметан); алкилариловые R-O-Ar ( метилфениловый эфир, метоксибензол); диариловые Ar-O-Ar ( дифениловый эфир).
ПРОСТЫЕ ЭФИРЫ Номенклатура и классификация I. Нециклические: диалкиловые эфиры R-O-R (напр. , CH 3 -O-CH 3 диметиловый эфир, метоксиметан); алкилариловые R-O-Ar ( метилфениловый эфир, метоксибензол); диариловые Ar-O-Ar ( дифениловый эфир).
 Большинство из них - жидкости, являются хорошими растворителями, инертными к проводимым в них реакциям. Нециклические эфиры в воде практически нерастворимы, тогда как циклические часто смешиваются с ней в любых соотношениях. Согласно тривиальной номенклатуре, простые эфиры называют по заместителям, связанным с атомом кислорода, добавляя слово «эфир» :
Большинство из них - жидкости, являются хорошими растворителями, инертными к проводимым в них реакциям. Нециклические эфиры в воде практически нерастворимы, тогда как циклические часто смешиваются с ней в любых соотношениях. Согласно тривиальной номенклатуре, простые эфиры называют по заместителям, связанным с атомом кислорода, добавляя слово «эфир» :
 По номенклатуре ИЮПАК эфиры рассматривают как алкоксиалканы. Корень названия определяет наиболее длинная алкильная группа: Простые эфиры относятся к числу малореакционноспособных веществ и стабильны по отношению ко многим реагентам (металлоорганическим соединениям и др. ), но они чувствительны по отношению к кислороду и легко образуют взрывчатые гидропероксиды, которые являются причиной взрыва при неосторожном обращении.
По номенклатуре ИЮПАК эфиры рассматривают как алкоксиалканы. Корень названия определяет наиболее длинная алкильная группа: Простые эфиры относятся к числу малореакционноспособных веществ и стабильны по отношению ко многим реагентам (металлоорганическим соединениям и др. ), но они чувствительны по отношению к кислороду и легко образуют взрывчатые гидропероксиды, которые являются причиной взрыва при неосторожном обращении.
 ПОЛУЧЕНИЕ ПРОСТЫХ ЭФИРОВ Существует три общих метода получения простых эфиров: межмолекулярная дегидратация спиртов, алкоксимеркурирование алкенов и реакция А. Вильямсона. 1. Межмолекулярная дегидратация спиртов Способ пригоден для получения симметричных простых эфиров из неразветвленных первичных спиртов:
ПОЛУЧЕНИЕ ПРОСТЫХ ЭФИРОВ Существует три общих метода получения простых эфиров: межмолекулярная дегидратация спиртов, алкоксимеркурирование алкенов и реакция А. Вильямсона. 1. Межмолекулярная дегидратация спиртов Способ пригоден для получения симметричных простых эфиров из неразветвленных первичных спиртов:
 2. Алкоксимеркурирование алкенов по существу аналогично оксимеркурированию, единственное различие состоит в том, что роль «внешнего» нуклеофильного агента выполняет спирт, который используется в качестве растворителя.
2. Алкоксимеркурирование алкенов по существу аналогично оксимеркурированию, единственное различие состоит в том, что роль «внешнего» нуклеофильного агента выполняет спирт, который используется в качестве растворителя.
 3. Синтез простых эфиров по Вильямсону Наиболее простой метод получения простых эфиров заключается во взаимодействии алкоголятов щелочных металлов с алкилгалогенидами или алкилсульфонатами. Эта реакция была открыта А. Вильямсоном в 1852 году и до сих пор остается наиболее общим способом получения простых эфиров:
3. Синтез простых эфиров по Вильямсону Наиболее простой метод получения простых эфиров заключается во взаимодействии алкоголятов щелочных металлов с алкилгалогенидами или алкилсульфонатами. Эта реакция была открыта А. Вильямсоном в 1852 году и до сих пор остается наиболее общим способом получения простых эфиров:
 Реакция Вильямсона пригодна для синтеза как симметричных, так и несимметричных простых эфиров: Получение простых эфиров по Вильямсону представляет собой обычную реакцию бимолекулярного нуклеофильного замещения у насыщенного атома углерода SN 2 с помощью алкоксид- или феноксид-ионов. Эта старая реакция неожиданно обрела второе рождение после открытия нового класса простых эфиров, так называемых краун-полиэфиров.
Реакция Вильямсона пригодна для синтеза как симметричных, так и несимметричных простых эфиров: Получение простых эфиров по Вильямсону представляет собой обычную реакцию бимолекулярного нуклеофильного замещения у насыщенного атома углерода SN 2 с помощью алкоксид- или феноксид-ионов. Эта старая реакция неожиданно обрела второе рождение после открытия нового класса простых эфиров, так называемых краун-полиэфиров.
 Краун-полиэфирами называют макроциклические полиэфиры, содержащие несколько атомов кислорода в цикле. Все краунполиэфиры характеризуются регулярной структурой, где каждые два атома кислорода в цикле связаны посредством двух метиленовых звеньев, т. е. формально краун-полиэфиры можно рассматривать как продукты циклоолигомеризации окиси этилена. В названиях краунполиэфиров первая цифра указывает на размер цикла, а вторая определяет число атомов кислорода в цикле.
Краун-полиэфирами называют макроциклические полиэфиры, содержащие несколько атомов кислорода в цикле. Все краунполиэфиры характеризуются регулярной структурой, где каждые два атома кислорода в цикле связаны посредством двух метиленовых звеньев, т. е. формально краун-полиэфиры можно рассматривать как продукты циклоолигомеризации окиси этилена. В названиях краунполиэфиров первая цифра указывает на размер цикла, а вторая определяет число атомов кислорода в цикле.
 Первый краун-полиэфир дибензо-18 -краун-6 был получен в 1967 г. с помощью реакции Вильямсона между динатриевой солью пирокатехина и бис-(2 -хлорэтиловым) эфиром:
Первый краун-полиэфир дибензо-18 -краун-6 был получен в 1967 г. с помощью реакции Вильямсона между динатриевой солью пирокатехина и бис-(2 -хлорэтиловым) эфиром:
 Краун-полиэфиры образуют стабильные комплексы с катионами непереходных и переходных металлов. Стабильность этих комплексов зависит от соответствия диаметра катиона размеру полости кольца, а также от координационного числа катиона металла. Комплексообразование краун-полиэфиров, их сернистых и азотных аналогов, а также полициклических краун-соединений - так называемых криптандов - с катионами металлов составляет интересный самостоятельный раздел современной аналитической химии:
Краун-полиэфиры образуют стабильные комплексы с катионами непереходных и переходных металлов. Стабильность этих комплексов зависит от соответствия диаметра катиона размеру полости кольца, а также от координационного числа катиона металла. Комплексообразование краун-полиэфиров, их сернистых и азотных аналогов, а также полициклических краун-соединений - так называемых криптандов - с катионами металлов составляет интересный самостоятельный раздел современной аналитической химии:
 Химические свойства Характерна низкая реакционная способность. 1. Эфиры как жесткие основания Льюиса образуют очень прочные комплексы с жесткими кислотами Льюиса - BF 3, Аl. Вr 3, Аl. R 3, Sb. Cl 5, Sb. F 5, Sn. Cl 4, Zn. Cl 2 и т. д. состава 1: 1 или 1: 2:
Химические свойства Характерна низкая реакционная способность. 1. Эфиры как жесткие основания Льюиса образуют очень прочные комплексы с жесткими кислотами Льюиса - BF 3, Аl. Вr 3, Аl. R 3, Sb. Cl 5, Sb. F 5, Sn. Cl 4, Zn. Cl 2 и т. д. состава 1: 1 или 1: 2:
 Эфиры образуют соли триалкилоксония при взаимодействии с очень сильными алкилирующими агентами: На способности простых эфиров давать соли оксония основаны способы расщепления простых эфиров под действием бромистоводородной или йодистоводородной кислоты, а также тригалогенидов бора.
Эфиры образуют соли триалкилоксония при взаимодействии с очень сильными алкилирующими агентами: На способности простых эфиров давать соли оксония основаны способы расщепления простых эфиров под действием бромистоводородной или йодистоводородной кислоты, а также тригалогенидов бора.
 2. Кислотное расщепление простых эфиров Простые эфиры расщепляются при нагревании до 120 -150 °С с концентрированными (48%) водными растворами НВr или HI. CH 3 -O-CH 3 + HJ CH 3 J + CH 3 ОН - типичный случай реакции нуклеофильного замещения у насыщенного атома углерода.
2. Кислотное расщепление простых эфиров Простые эфиры расщепляются при нагревании до 120 -150 °С с концентрированными (48%) водными растворами НВr или HI. CH 3 -O-CH 3 + HJ CH 3 J + CH 3 ОН - типичный случай реакции нуклеофильного замещения у насыщенного атома углерода.
 Если эфир содержит первичные или вторичные алкильные группы, реализуется SN 2 -механизм, в котором бромид- или йодид -ион атакует протонированную форму эфира по менее замещенному атому углерода: В том случае, когда эфир содержит первичную и вторичную алкильные группы, расщепление отличается высокой региоселективностью и, как правило, образуется только один из двух возможных спиртов (вторичный) и только первичный алкилгалогенид.
Если эфир содержит первичные или вторичные алкильные группы, реализуется SN 2 -механизм, в котором бромид- или йодид -ион атакует протонированную форму эфира по менее замещенному атому углерода: В том случае, когда эфир содержит первичную и вторичную алкильные группы, расщепление отличается высокой региоселективностью и, как правило, образуется только один из двух возможных спиртов (вторичный) и только первичный алкилгалогенид.
 3. РАДИКАЛЬНЫЕ РЕАКЦИИ ПРОСТЫХ ЭФИРОВ Подобно алканам, простые эфиры вступают в реакцию радикального галогенирования, однако галогенирование эфиров отличается высокой региоселективностью и осуществляется в αположение по отношению к атому кислорода. 4. Образование перекисей СН 3 -СН 2 -О-СН 2 -СН 3 Образующиеся соединения взрывоопасны, поэтому простые эфиры рекомендуется хранить под щелочью.
3. РАДИКАЛЬНЫЕ РЕАКЦИИ ПРОСТЫХ ЭФИРОВ Подобно алканам, простые эфиры вступают в реакцию радикального галогенирования, однако галогенирование эфиров отличается высокой региоселективностью и осуществляется в αположение по отношению к атому кислорода. 4. Образование перекисей СН 3 -СН 2 -О-СН 2 -СН 3 Образующиеся соединения взрывоопасны, поэтому простые эфиры рекомендуется хранить под щелочью.
![ДИОКСИНЫ Диоксины –тривиальное название полихлорпроизводных дибензо[b, e]-1, 4 -диоксина. Название происходит от сокращённого названия](https://present5.com/presentation/-32391069_145318438/image-98.jpg "ДИОКСИНЫ Диоксины –тривиальное название полихлорпроизводных дибензо[b, e]-1, 4 -диоксина. Название происходит от сокращённого названия") ДИОКСИНЫ Диоксины –тривиальное название полихлорпроизводных дибензо[b, e]-1, 4 -диоксина. Название происходит от сокращённого названия тетрахлорпроизводного – 2, 3, 7, 8 тетрахлордибензо[b, е]-1, 4 -диоксина; соединения с другими заместителями – галогенидами – также относятся к диоксинам. Являются кумулятивными ядами и относятся к группе опасных ксенобиотиков.
ДИОКСИНЫ Диоксины –тривиальное название полихлорпроизводных дибензо[b, e]-1, 4 -диоксина. Название происходит от сокращённого названия тетрахлорпроизводного – 2, 3, 7, 8 тетрахлордибензо[b, е]-1, 4 -диоксина; соединения с другими заместителями – галогенидами – также относятся к диоксинам. Являются кумулятивными ядами и относятся к группе опасных ксенобиотиков.
 2, 3, 7, 8 -тетрахлордибензо-пара-диоксин - самое смертоносное вещество из 75 известных диоксинов. Как яд оно в 150 000 раз сильнее цианида. Является самым ядовитым искусственно синтезированным веществом на Земле. Диоксины – это глобальные экотоксиканты, обладающие мощным мутагенным, иммунодепрессантным, канцерогенным, тератогенным и эмбриотоксическим действием. Они слабо расщепляются и накапливаются как в организме человека, так и в биосфере планеты, включая воздух, воду, пищу. Величина летальной дозы для этих веществ достигает 10− 6 г на 1 кг живого веса, что существенно меньше аналогичной величины для некоторых боевых отравляющих веществ, например, для зомана, зарина и табуна (порядка 10− 3 г/кг). Диоксины образуются в качестве побочного продукта при производствегербицидов хлорфенольного ряда (прежде всего, производных 2, 4 -дихлорфеноксиуксусной и 2, 4, 5 трихлорфеноксиуксусной кислот, а также их эфиров).
2, 3, 7, 8 -тетрахлордибензо-пара-диоксин - самое смертоносное вещество из 75 известных диоксинов. Как яд оно в 150 000 раз сильнее цианида. Является самым ядовитым искусственно синтезированным веществом на Земле. Диоксины – это глобальные экотоксиканты, обладающие мощным мутагенным, иммунодепрессантным, канцерогенным, тератогенным и эмбриотоксическим действием. Они слабо расщепляются и накапливаются как в организме человека, так и в биосфере планеты, включая воздух, воду, пищу. Величина летальной дозы для этих веществ достигает 10− 6 г на 1 кг живого веса, что существенно меньше аналогичной величины для некоторых боевых отравляющих веществ, например, для зомана, зарина и табуна (порядка 10− 3 г/кг). Диоксины образуются в качестве побочного продукта при производствегербицидов хлорфенольного ряда (прежде всего, производных 2, 4 -дихлорфеноксиуксусной и 2, 4, 5 трихлорфеноксиуксусной кислот, а также их эфиров).
 Так, например, производство 2, 4, 5 -трихлорфеноксиуксусной кислоты включает последовательные стадии гидролиза тетрахлорбензола в метанольном растворе щелочью с получением 2, 4, 5 -трихлорфенолята натрия и последующее алкилирование 2, 4, 5 -трихлорфенолята натрия трихлоруксусной кислотой; 2, 3, 7, 8 тетрахлордибензо-пара-диоксин образуется на обеих стадиях при самоконденсации 2, 4, 5 -трихлорфенолята натрия:
Так, например, производство 2, 4, 5 -трихлорфеноксиуксусной кислоты включает последовательные стадии гидролиза тетрахлорбензола в метанольном растворе щелочью с получением 2, 4, 5 -трихлорфенолята натрия и последующее алкилирование 2, 4, 5 -трихлорфенолята натрия трихлоруксусной кислотой; 2, 3, 7, 8 тетрахлордибензо-пара-диоксин образуется на обеих стадиях при самоконденсации 2, 4, 5 -трихлорфенолята натрия:
 Основные причины эмиссии диоксинов в биосферу – прежде всего использование высокотемпературных технологий хлорирования и переработки хлорорганических веществ и, особенно, сжигание отходов производства. Наличие в уничтожаемом мусоре повсеместно распространённого поливинилхлорида и других полимеров, различных соединений хлора способствует образованию в дымовых газах диоксинов. Другой источник опасности – целлюлозно-бумажная промышленность. Отбеливание целлюлозной пульпы хлором сопровождается образованием диоксинов и ряда других опасных хлорорганических веществ.
Основные причины эмиссии диоксинов в биосферу – прежде всего использование высокотемпературных технологий хлорирования и переработки хлорорганических веществ и, особенно, сжигание отходов производства. Наличие в уничтожаемом мусоре повсеместно распространённого поливинилхлорида и других полимеров, различных соединений хлора способствует образованию в дымовых газах диоксинов. Другой источник опасности – целлюлозно-бумажная промышленность. Отбеливание целлюлозной пульпы хлором сопровождается образованием диоксинов и ряда других опасных хлорорганических веществ.
 СЕРУСОДЕРЖАЩИЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ Содержат связь С-S в составе различных функциональных групп. Атом серы имеет электронное строение: 0 3 s 2 3 p 4 2 s 2 2 p 6 1 s 2 На его внешнем электронном уровне 6 электронов: sэлектронная пара, р-электронная пара и два неспаренных рэлектрона. При образовании химических связей по одному электрону с 3 p- и 3 s- уровней могут последовательно переходить на 3 d-орбитали, тем самым обеспечивая валентности атома серы 2, 4 и 6.
СЕРУСОДЕРЖАЩИЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ Содержат связь С-S в составе различных функциональных групп. Атом серы имеет электронное строение: 0 3 s 2 3 p 4 2 s 2 2 p 6 1 s 2 На его внешнем электронном уровне 6 электронов: sэлектронная пара, р-электронная пара и два неспаренных рэлектрона. При образовании химических связей по одному электрону с 3 p- и 3 s- уровней могут последовательно переходить на 3 d-орбитали, тем самым обеспечивая валентности атома серы 2, 4 и 6.
 Номенклатура и классификация В органических соединениях атом серы может находиться в разных степенях окисления. 1. Сульфоновые кислоты (сульфокислоты), производные сульфоновых кислот (галогенангидриды RSO 2 X, сложные эфиры R-SO 2 -O-R’, соли R-SO 3 Na, амиды R-SO 2 NH 2 и др. ) и сульфоны:
Номенклатура и классификация В органических соединениях атом серы может находиться в разных степенях окисления. 1. Сульфоновые кислоты (сульфокислоты), производные сульфоновых кислот (галогенангидриды RSO 2 X, сложные эфиры R-SO 2 -O-R’, соли R-SO 3 Na, амиды R-SO 2 NH 2 и др. ) и сульфоны:
. Например:") Сера в этих соединениях находится в степени окисления +6 (органические аналоги серной кислоты). Например: метансульфоновая бензолсульфоновая 2 -метилпропан-2 кислота (метан кислота (бензол 1, 4 -бензолди сульфонат натрия сульфокислота) сульфокислота (соль сульфокислоты)
Сера в этих соединениях находится в степени окисления +6 (органические аналоги серной кислоты). Например: метансульфоновая бензолсульфоновая 2 -метилпропан-2 кислота (метан кислота (бензол 1, 4 -бензолди сульфонат натрия сульфокислота) сульфокислота (соль сульфокислоты)
 метилэтан сульфонат (метиловый эфир этансульфоновой кислоты) метилэтил п-толуол") этансульфонил хлорид (хлорангидрид этансульфоновой кислоты) метилэтан сульфонат (метиловый эфир этансульфоновой кислоты) метилэтил п-толуол сульфонамид (амид птолуолсульфоновой кислоты)
этансульфонил хлорид (хлорангидрид этансульфоновой кислоты) метилэтан сульфонат (метиловый эфир этансульфоновой кислоты) метилэтил п-толуол сульфонамид (амид птолуолсульфоновой кислоты)
 2. Сульфиновые кислоты, производные сульфиновых кислот и сульфоксиды как органические аналоги сернистой кислоты. Сера в этих соединениях находится в степени окисления +4: Например: метансульфиновая кислота диметилсульфоксид
2. Сульфиновые кислоты, производные сульфиновых кислот и сульфоксиды как органические аналоги сернистой кислоты. Сера в этих соединениях находится в степени окисления +4: Например: метансульфиновая кислота диметилсульфоксид
: Нестабильны, изучены мало. пропен-1 -сульфеновая кислота") 3. Сульфеновые кислоты и их производные (S 2+): Нестабильны, изучены мало. пропен-1 -сульфеновая кислота (определяет слезоточивое действие лука) 4. Тиолы (меркаптаны), или органические аналоги сернистой кислоты (S 2 -): Например, метантиол 2 -бутантиол
3. Сульфеновые кислоты и их производные (S 2+): Нестабильны, изучены мало. пропен-1 -сульфеновая кислота (определяет слезоточивое действие лука) 4. Тиолы (меркаптаны), или органические аналоги сернистой кислоты (S 2 -): Например, метантиол 2 -бутантиол
 5. Органические сульфиды, дисульфиды, полисульфиды По номенклатуре ИЮПАК сульфиды называются алкилтиоалканами (принцип построения названий аналогичен названиям простых эфиров): 1 2 3 4 1 -(изопропилтио)бутан В соответствии с общей номенклатурой перед словом «сульфид» дается название двух алкильных групп (изопропилбутилсульфид).
5. Органические сульфиды, дисульфиды, полисульфиды По номенклатуре ИЮПАК сульфиды называются алкилтиоалканами (принцип построения названий аналогичен названиям простых эфиров): 1 2 3 4 1 -(изопропилтио)бутан В соответствии с общей номенклатурой перед словом «сульфид» дается название двух алкильных групп (изопропилбутилсульфид).
 Серусодержащие органические соединения получаются в окислительно-восстановительных процессах, однако именно реакции окисления распространены более широко:
Серусодержащие органические соединения получаются в окислительно-восстановительных процессах, однако именно реакции окисления распространены более широко:
 ТИОЛЫ И СУЛЬФИДЫ НОМЕНКЛАТУРА И КЛАССИФИКАЦИЯ Рассматриваются как производные спиртов и эфиров. Тиолы в номенклатуре ИЮПАК имеют окончание тиол: Старинное название сернистых аналогов спиртов - меркаптаны и SHгруппы как меркапто- теперь редко употребляется. Сульфиды называют аналогично простым эфирам. В соответствии с общей номенклатурой перед словом сульфид дается название двух алкильных или арильных групп, например:
ТИОЛЫ И СУЛЬФИДЫ НОМЕНКЛАТУРА И КЛАССИФИКАЦИЯ Рассматриваются как производные спиртов и эфиров. Тиолы в номенклатуре ИЮПАК имеют окончание тиол: Старинное название сернистых аналогов спиртов - меркаптаны и SHгруппы как меркапто- теперь редко употребляется. Сульфиды называют аналогично простым эфирам. В соответствии с общей номенклатурой перед словом сульфид дается название двух алкильных или арильных групп, например:
 СТРОЕНИЕ И ФИЗИЧЕСКИЕ СВОЙСТВА Тиолы – вещества с резким неприятным запахом. Угол C-S -H изменяется в пределах 100– 108 о. Связи C-S и S-H полярны, дипольные моменты алкантиолов составляют 1, 3 -1, 5 D. Молекулы тиолов могут образовывать друг с другом водородные связи. По строению метантиол напоминает метанол, но связь C— S значительно длиннее связи С—О.
СТРОЕНИЕ И ФИЗИЧЕСКИЕ СВОЙСТВА Тиолы – вещества с резким неприятным запахом. Угол C-S -H изменяется в пределах 100– 108 о. Связи C-S и S-H полярны, дипольные моменты алкантиолов составляют 1, 3 -1, 5 D. Молекулы тиолов могут образовывать друг с другом водородные связи. По строению метантиол напоминает метанол, но связь C— S значительно длиннее связи С—О.
 Тиолы проявляют свойства более сильных кислот, чем спирты, подобно тому как H 2 S (р. Ka=7, 05) диссоциирован сильнее, чем вода. Для тиолов р. Ka изменяется в интервале 9, 511, т. е. гидроксид-ион превращает тиолы нацело в тиолат-ион: Связь S-H значительно менее полярна, чем связь O-Н, и тиолы связаны между собой очень слабой межмолекулярной водородной связью в отличие от спиртов. Это выражается в более низкой температуре кипения тиолов. Так, например, этантиол кипит при 37 o. С, тогда как этанол - при 78 o. С.
Тиолы проявляют свойства более сильных кислот, чем спирты, подобно тому как H 2 S (р. Ka=7, 05) диссоциирован сильнее, чем вода. Для тиолов р. Ka изменяется в интервале 9, 511, т. е. гидроксид-ион превращает тиолы нацело в тиолат-ион: Связь S-H значительно менее полярна, чем связь O-Н, и тиолы связаны между собой очень слабой межмолекулярной водородной связью в отличие от спиртов. Это выражается в более низкой температуре кипения тиолов. Так, например, этантиол кипит при 37 o. С, тогда как этанол - при 78 o. С.
 ПОЛУЧЕНИЕ 1. Самый старый метод получения тиолов основан на реакции бимолекулярного нуклеофильного замещения галогенид-иона в первичных и вторичных алкилгалогенидах под действием гидросульфид-иона: 2. Современный метод синтеза тиолов заключается во взаимодействии алкилгалогенидов или алкилсульфонатов с тиомочевиной: Na. OH выход 83%
ПОЛУЧЕНИЕ 1. Самый старый метод получения тиолов основан на реакции бимолекулярного нуклеофильного замещения галогенид-иона в первичных и вторичных алкилгалогенидах под действием гидросульфид-иона: 2. Современный метод синтеза тиолов заключается во взаимодействии алкилгалогенидов или алкилсульфонатов с тиомочевиной: Na. OH выход 83%
 с разрывом связи SH и") СВОЙСТВА Реакционные центры: Характерны 2 основные группы реакций: а) с разрывом связи SH и б) нуклеофильные реакции с участием атома серы. 1. Кислотные свойства R-S-H + : B R-S- + HB+, где : B – основание Тиолы проявляют более выраженные кислотные свойства, чем спирты R-OH.
СВОЙСТВА Реакционные центры: Характерны 2 основные группы реакций: а) с разрывом связи SH и б) нуклеофильные реакции с участием атома серы. 1. Кислотные свойства R-S-H + : B R-S- + HB+, где : B – основание Тиолы проявляют более выраженные кислотные свойства, чем спирты R-OH.
 2. Окисление: Окисление тиолов резко отличается от окисления спиртов. В зависимости от природы окислителя продуктами окисления тиолов являются дисульфиды R-S-S-R, сульфиновые RSO 2 H или сульфоновые RSO 3 H кислоты. При действии таких окислителей, как йод, бром, пероксид водорода, Мn. О 3, тиолы окисляются до дисульфидов:
2. Окисление: Окисление тиолов резко отличается от окисления спиртов. В зависимости от природы окислителя продуктами окисления тиолов являются дисульфиды R-S-S-R, сульфиновые RSO 2 H или сульфоновые RSO 3 H кислоты. При действии таких окислителей, как йод, бром, пероксид водорода, Мn. О 3, тиолы окисляются до дисульфидов:
 Перкислоты, например мета-хлорпербензойная кислота, в исключительно мягких условиях окисляют тиолы до сульфиновых кислот: Сильные окислители - азотная кислота или перманганат калия - окисляют тиолы до сульфоновых кислот (продуктов исчерпывающего окисления органических соединений серы):
Перкислоты, например мета-хлорпербензойная кислота, в исключительно мягких условиях окисляют тиолы до сульфиновых кислот: Сильные окислители - азотная кислота или перманганат калия - окисляют тиолы до сульфоновых кислот (продуктов исчерпывающего окисления органических соединений серы):
 Анионы тиолов вступают в") Сульфиды окисляются последовательно до сульфоксидов и далее до сульфонов: б) Анионы тиолов вступают в реакцию Вильямсона, приводящую к получению тиоэфиров (сульфидов): Тиолят-ионы являются более сильными нуклеофильными агентами, чем алкоголят-ионы, и скорость образования тиоэфиров в 103 -104 раза превышает скорость реакции для их кислородных аналогов.
Сульфиды окисляются последовательно до сульфоксидов и далее до сульфонов: б) Анионы тиолов вступают в реакцию Вильямсона, приводящую к получению тиоэфиров (сульфидов): Тиолят-ионы являются более сильными нуклеофильными агентами, чем алкоголят-ионы, и скорость образования тиоэфиров в 103 -104 раза превышает скорость реакции для их кислородных аналогов.
 Сульфиды могут быть также получены в результате прямого взаимодействия сульфида натрия с двумя молями алкилирующего агента: У сульфидов остается единственный активный реакционный центр – неподеленная пара электронов атома S: 1. Являются очень слабыми основаниями: р. КВН+ = -5, 25
Сульфиды могут быть также получены в результате прямого взаимодействия сульфида натрия с двумя молями алкилирующего агента: У сульфидов остается единственный активный реакционный центр – неподеленная пара электронов атома S: 1. Являются очень слабыми основаниями: р. КВН+ = -5, 25
 2. При алкилировании образуют соли сульфония: 3. Легко окисляются с образованием сульфоксидов и сульфонов.
2. При алкилировании образуют соли сульфония: 3. Легко окисляются с образованием сульфоксидов и сульфонов.
 СУЛЬФОКСИДЫ СТРОЕНИЕ МОЛЕКУЛ Дипольные моменты сульфоксидов R 2 S=O довольно велики (3, 8 – 4, 0 D), поэтому связь S=O в них часто считают ионизированной S+–O-. Низшие члены этого ряда являются хорошими растворителями как органических, так и неорганических соединений (сверхрастворители – диметилсульфоксид, сульфолан). Сульфоксиды принадлежат к числу соединений с трехкоординированным атомом серы. Природа связи серы и кислорода в сульфоксидах в течение длительного времени была предметом дискуссий и острых споров. Для сульфоксидной группы обычно используют два обозначения: S+—O-, которое подчеркивает диполярную, донорно-акцепторную природу связи серы и кислорода, и S=O, которое указывает на dπ*-pπ-взаимодействие вакантной d-орбитали серы и неподеленной пары p-электронов кислорода. Длина связи серы и кислорода в сульфоксидах 1, 47 Å формально согласуется с двойной связью, поскольку одинарная связь S-O имеет длину 1, 69 Å.
СУЛЬФОКСИДЫ СТРОЕНИЕ МОЛЕКУЛ Дипольные моменты сульфоксидов R 2 S=O довольно велики (3, 8 – 4, 0 D), поэтому связь S=O в них часто считают ионизированной S+–O-. Низшие члены этого ряда являются хорошими растворителями как органических, так и неорганических соединений (сверхрастворители – диметилсульфоксид, сульфолан). Сульфоксиды принадлежат к числу соединений с трехкоординированным атомом серы. Природа связи серы и кислорода в сульфоксидах в течение длительного времени была предметом дискуссий и острых споров. Для сульфоксидной группы обычно используют два обозначения: S+—O-, которое подчеркивает диполярную, донорно-акцепторную природу связи серы и кислорода, и S=O, которое указывает на dπ*-pπ-взаимодействие вакантной d-орбитали серы и неподеленной пары p-электронов кислорода. Длина связи серы и кислорода в сульфоксидах 1, 47 Å формально согласуется с двойной связью, поскольку одинарная связь S-O имеет длину 1, 69 Å.
 Несимметричные cульфоксиды хиральны, и роль четвертого заместителя выполняет неподеленная пара электронов атома серы: Сульфоксиды относятся к незаряженным амбидентным нуклеофилам. Термин «амбидентный нуклеофил» применяется для нейтральных соединений и анионов, содержащих два нуклеофильных центра. В сульфоксидах это атомы серы и кислорода.
Несимметричные cульфоксиды хиральны, и роль четвертого заместителя выполняет неподеленная пара электронов атома серы: Сульфоксиды относятся к незаряженным амбидентным нуклеофилам. Термин «амбидентный нуклеофил» применяется для нейтральных соединений и анионов, содержащих два нуклеофильных центра. В сульфоксидах это атомы серы и кислорода.
 Получают окислением сульфидов разбавленной азотной кислотой или оксидами азота: R-S-R’ Реакционные центры: Сульфоксиды, как и сульфоны, содержат сильные электроноакцепторные группы SO и SO 2. Эти группы за счет сильного -I-эффекта вызывают поляризацию С-Н-связи при α -углеродном атоме, и водород в виде протона отщепляется при действии очень сильных оснований, таких, как гидрид натрия, трет-бутилат калия или бутиллитий.
Получают окислением сульфидов разбавленной азотной кислотой или оксидами азота: R-S-R’ Реакционные центры: Сульфоксиды, как и сульфоны, содержат сильные электроноакцепторные группы SO и SO 2. Эти группы за счет сильного -I-эффекта вызывают поляризацию С-Н-связи при α -углеродном атоме, и водород в виде протона отщепляется при действии очень сильных оснований, таких, как гидрид натрия, трет-бутилат калия или бутиллитий.
 Т. о. проявляют свойства слабых оснований и очень слабых СН-кислот:
Т. о. проявляют свойства слабых оснований и очень слабых СН-кислот:
 СУЛЬФОНОВЫЕ КИСЛОТЫ Сульфоновые кислоты и сульфоны можно рассматривать как органические производные серной кислоты, в молекуле которой одна или обе гидроксильные группы замещены углеводородными остатками. Производными сульфоновых кислот являются их эфиры , амиды , хлорангидриды Сульфоновые кислоты и их производные – бесцветные вещества, большинство из них кристаллические, растворимые в воде.
СУЛЬФОНОВЫЕ КИСЛОТЫ Сульфоновые кислоты и сульфоны можно рассматривать как органические производные серной кислоты, в молекуле которой одна или обе гидроксильные группы замещены углеводородными остатками. Производными сульфоновых кислот являются их эфиры , амиды , хлорангидриды Сульфоновые кислоты и их производные – бесцветные вещества, большинство из них кристаллические, растворимые в воде.
 СТРОЕНИЕ МОЛЕКУЛ Сульфонат-ион имеет тетраэдрическое строение, углы между связями 108 -110 о: Все три связи S-O одинаковые, и отрицательный заряд делокализован по всем трем кислородным атомам.
СТРОЕНИЕ МОЛЕКУЛ Сульфонат-ион имеет тетраэдрическое строение, углы между связями 108 -110 о: Все три связи S-O одинаковые, и отрицательный заряд делокализован по всем трем кислородным атомам.
 Какова природа связи S=O, является ли она чистой семиполярной связью S+-O- или полярной двойной связью? Формально ее структуру можно изобразить с помощью семиполярных связей, например: В то же время атом серы имеет незаполненные 3 d-орбитали, которые могут быть использованы для делокализации электронных пар кислородных атомов. Так образуется полярная двойная связь: , где = 0, 5 – 0, 6.
Какова природа связи S=O, является ли она чистой семиполярной связью S+-O- или полярной двойной связью? Формально ее структуру можно изобразить с помощью семиполярных связей, например: В то же время атом серы имеет незаполненные 3 d-орбитали, которые могут быть использованы для делокализации электронных пар кислородных атомов. Так образуется полярная двойная связь: , где = 0, 5 – 0, 6.
 Это вытекает из значений дипольных моментов связей S=O (µ 10 10 -30 Кл м или 3 D). Если бы связь была чисто семиполярной, дипольный момент был бы равен 20 10 -30 - 23 10 -30 Кл м ( 6 -7 D). Группа -SO 2 является сильным электроноакцептором, проявляет –J и –С эффекты.
Это вытекает из значений дипольных моментов связей S=O (µ 10 10 -30 Кл м или 3 D). Если бы связь была чисто семиполярной, дипольный момент был бы равен 20 10 -30 - 23 10 -30 Кл м ( 6 -7 D). Группа -SO 2 является сильным электроноакцептором, проявляет –J и –С эффекты.
 Сульфирование аренов концентрированной серной кислотой или олеумом (H 2 SO") R-SO 3 H; 2) Сульфирование аренов концентрированной серной кислотой или олеумом (H 2 SO 4 + SO 3) при нагревании 2 + 2 H 2 SO 4 + (п-толуолсульфокислотата) (о-толуолсульфокислота) + 2 H 2 O
R-SO 3 H; 2) Сульфирование аренов концентрированной серной кислотой или олеумом (H 2 SO 4 + SO 3) при нагревании 2 + 2 H 2 SO 4 + (п-толуолсульфокислотата) (о-толуолсульфокислота) + 2 H 2 O
 Сульфонилхлориды как ароматические так и неароматические получают по реакции сульфохлорирования: алканов: R-H аренов:") 3) Сульфонилхлориды как ароматические так и неароматические получают по реакции сульфохлорирования: алканов: R-H аренов: R-SO 2 Сl + 2 Cl. SO 3 H + HCl + H 2 SO 4 избыток хлорсульфоновой кислоты Один из методов получения хлорангидридов сульфокислот (сульфохлоридов) заключается в обработке сухой натриевой соли тионилхлоридом в ДМФА:
3) Сульфонилхлориды как ароматические так и неароматические получают по реакции сульфохлорирования: алканов: R-H аренов: R-SO 2 Сl + 2 Cl. SO 3 H + HCl + H 2 SO 4 избыток хлорсульфоновой кислоты Один из методов получения хлорангидридов сульфокислот (сульфохлоридов) заключается в обработке сухой натриевой соли тионилхлоридом в ДМФА:
 Из сульфонилхлоридов получают другие производные сульфоновых кислот: А также исходные сульфоновые кислоты: R-SO 2 Сl + Н 2 О R-SO 2 ОН + HCl
Из сульфонилхлоридов получают другие производные сульфоновых кислот: А также исходные сульфоновые кислоты: R-SO 2 Сl + Н 2 О R-SO 2 ОН + HCl
 Химические свойства Реакционные центры: 1. Сульфоновые кислоты являются сильными кислотами, в водных растворах ионизированы: R-SO 3 H + H 2 O R-SO 3 - + H 3 O+ По кислотности сульфоновые кислоты сравнимы с серной кислотой. Наиболее сильной из них является трифторметансульфоновая кислота CF 3 SO 3 H.
Химические свойства Реакционные центры: 1. Сульфоновые кислоты являются сильными кислотами, в водных растворах ионизированы: R-SO 3 H + H 2 O R-SO 3 - + H 3 O+ По кислотности сульфоновые кислоты сравнимы с серной кислотой. Наиболее сильной из них является трифторметансульфоновая кислота CF 3 SO 3 H.
 Эти кислоты образуют устойчивые соли. В большинстве случаев натриевые соли осаждаются из водных растворов сульфоновых кислот прибавлением хлорида натрия до насыщения ( «высаливание» сульфоновых кислот): Высаливание сульфоновых кислот часто применяют как способ их выделения из водного раствора. Синтетические моющие средства содержат смесь солей различных сульфоновых кислот с числом углеродных атомов С 10 С 20.
Эти кислоты образуют устойчивые соли. В большинстве случаев натриевые соли осаждаются из водных растворов сульфоновых кислот прибавлением хлорида натрия до насыщения ( «высаливание» сульфоновых кислот): Высаливание сульфоновых кислот часто применяют как способ их выделения из водного раствора. Синтетические моющие средства содержат смесь солей различных сульфоновых кислот с числом углеродных атомов С 10 С 20.
 или нуклеофильное (б) замещение сульфогруппы: Ar-H + H 2 SO 4") 2. Электрофильное (а) или нуклеофильное (б) замещение сульфогруппы: Ar-H + H 2 SO 4 а) Ar-SO 3 H + H 2 O б) Ar-SO 3 H + 2 Na. OH tо. С Ar-OH + Na 2 SO 3 + H 2 O Сульфогруппа может быть заменена на нитрогруппу или галоген. Эти электрофильные реакции ипсо-замещения широко используются в органическом синтезе, например при получении пикриновой кислоты:
2. Электрофильное (а) или нуклеофильное (б) замещение сульфогруппы: Ar-H + H 2 SO 4 а) Ar-SO 3 H + H 2 O б) Ar-SO 3 H + 2 Na. OH tо. С Ar-OH + Na 2 SO 3 + H 2 O Сульфогруппа может быть заменена на нитрогруппу или галоген. Эти электрофильные реакции ипсо-замещения широко используются в органическом синтезе, например при получении пикриновой кислоты:
 сульфонилхлориды (галогенангидриды сульфоновых кислот) Связь S +-Cl - сильно полярна,") ПРОИЗВОДНЫЕ СУЛЬФОНОВЫХ КИСЛОТ а) сульфонилхлориды (галогенангидриды сульфоновых кислот) Связь S +-Cl - сильно полярна, поэтому сульфонилхлориды реакционноспособны и легко взаимодействуют с нуклеофилами. Из сульфонилхлоридов синтезируют другие производные сульфоновых кислот – эфиры, амиды, гидразиды и т. д. R-SO 2 -Cl + HNu: + B: R-SO 2 -Nu + HB+Cl-, где B: – основание
ПРОИЗВОДНЫЕ СУЛЬФОНОВЫХ КИСЛОТ а) сульфонилхлориды (галогенангидриды сульфоновых кислот) Связь S +-Cl - сильно полярна, поэтому сульфонилхлориды реакционноспособны и легко взаимодействуют с нуклеофилами. Из сульфонилхлоридов синтезируют другие производные сульфоновых кислот – эфиры, амиды, гидразиды и т. д. R-SO 2 -Cl + HNu: + B: R-SO 2 -Nu + HB+Cl-, где B: – основание
 сульфонамиды Слабые NH-кислоты, анион сульфонамида – нуклеофил, легко алкилируется и галогенируется:") б) сульфонамиды Слабые NH-кислоты, анион сульфонамида – нуклеофил, легко алкилируется и галогенируется:
б) сульфонамиды Слабые NH-кислоты, анион сульфонамида – нуклеофил, легко алкилируется и галогенируется:
 СУЛЬФОНЫ Соединения с двумя полярными двойными связями S +=O -, дипольный момент этой связи около 3 D. Группа SO 2 является сильным электроноакцептором, проявляет –I и, если атом S может взаимодействовать с системой сопряжения, –C-эффекты.
СУЛЬФОНЫ Соединения с двумя полярными двойными связями S +=O -, дипольный момент этой связи около 3 D. Группа SO 2 является сильным электроноакцептором, проявляет –I и, если атом S может взаимодействовать с системой сопряжения, –C-эффекты.
 Арилсульфоны получают по реакции сульфонилирования: + HCl Алкилсульфоны могут быть получены при окислении алкилсульфидов сильным окислителем. Сульфоны – инертные и термически стабильные соединения, являются слабыми СН-кислотами:
Арилсульфоны получают по реакции сульфонилирования: + HCl Алкилсульфоны могут быть получены при окислении алкилсульфидов сильным окислителем. Сульфоны – инертные и термически стабильные соединения, являются слабыми СН-кислотами:
 Содержат в своем составе карбонильную группу (альдегиды , кетоны") КАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ (АЛЬДЕГИДЫ И КЕТОНЫ) Содержат в своем составе карбонильную группу (альдегиды , кетоны ). Номенклатура и классификация Как и представители других классов функциональных производных углеводородов, классифицируются как насыщенные и ненасыщенные, линейные и циклические, ароматические и неароматические и т. д. В соответствии с номенклатурой ИЮПАК насыщенные альдегиды называют, начиная нумерацию главной цепи с атома углерода, входящего в состав карбонильной группы и прибавляя к названию суффикс –аль. Кетоны называют, прибавляя суффикс –он. Существует также рациональная и тривиальная номенклатура, в случае рациональной углеродная цепь обозначается буквами греческого алфавита, при этом Сатом карбонильной группы не отмечается.
КАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ (АЛЬДЕГИДЫ И КЕТОНЫ) Содержат в своем составе карбонильную группу (альдегиды , кетоны ). Номенклатура и классификация Как и представители других классов функциональных производных углеводородов, классифицируются как насыщенные и ненасыщенные, линейные и циклические, ароматические и неароматические и т. д. В соответствии с номенклатурой ИЮПАК насыщенные альдегиды называют, начиная нумерацию главной цепи с атома углерода, входящего в состав карбонильной группы и прибавляя к названию суффикс –аль. Кетоны называют, прибавляя суффикс –он. Существует также рациональная и тривиальная номенклатура, в случае рациональной углеродная цепь обозначается буквами греческого алфавита, при этом Сатом карбонильной группы не отмечается.
 Строение и электронные эффекты функциональной группы -I -I -С Атомы углерода и кислорода в карбонильной группе находятся в состоянии sp 2 -гибридизации. Углерод посредством sp 2 -гибридных орбиталей образует 3 -связи (одна из них - связь С–О), которые располагаются в одной плоскости под углом около 120° друг к другу. Одна из орбиталей атома кислорода участвует в -связи С–О, две другие содержат неподеленнные электронные пары.
Строение и электронные эффекты функциональной группы -I -I -С Атомы углерода и кислорода в карбонильной группе находятся в состоянии sp 2 -гибридизации. Углерод посредством sp 2 -гибридных орбиталей образует 3 -связи (одна из них - связь С–О), которые располагаются в одной плоскости под углом около 120° друг к другу. Одна из орбиталей атома кислорода участвует в -связи С–О, две другие содержат неподеленнные электронные пары.
 -Связь образована р-электронами атомов углерода и кислорода. Связь С=О сильно полярна. Ее дипольный момент (2, 6 -2, 8 D) значительно выше, чем у связи С–О в спиртах (0, 70 D). Электроны кратной связи С=О, в особенности более подвижные -электроны, смещены к электро-отрицательному атому кислорода, что приводит к появлению на нем частичного отрицательного заряда. Карбонильный углерод приобретает частичный положительный заряд. Поэтому карбонильная группа в альдегидах и кетонах является электроноакцепторным заместителем и может проявлять – I и – С-эффекты.
-Связь образована р-электронами атомов углерода и кислорода. Связь С=О сильно полярна. Ее дипольный момент (2, 6 -2, 8 D) значительно выше, чем у связи С–О в спиртах (0, 70 D). Электроны кратной связи С=О, в особенности более подвижные -электроны, смещены к электро-отрицательному атому кислорода, что приводит к появлению на нем частичного отрицательного заряда. Карбонильный углерод приобретает частичный положительный заряд. Поэтому карбонильная группа в альдегидах и кетонах является электроноакцепторным заместителем и может проявлять – I и – С-эффекты.
, а также некоторых ароматических") Физические свойства Являются жидкостями, за исключением метаналя и этаналя (газы), а также некоторых ароматических альдегидов и кетонов (твердые). Альдегиды и кетоны не образуют водородных связей, поэтому температуры их кипения ниже, чем у соответствующих спиртов, однако эти молекулы способны образовывать водородные связи с молекулами воды, поэтому растворимы в ней. Низшие члены ряда смешиваются с водой в любых соотношениях, однако уже растворимость бутаналя в воде составляет 7%. Ароматические соединения в воде малорастворимы.
Физические свойства Являются жидкостями, за исключением метаналя и этаналя (газы), а также некоторых ароматических альдегидов и кетонов (твердые). Альдегиды и кетоны не образуют водородных связей, поэтому температуры их кипения ниже, чем у соответствующих спиртов, однако эти молекулы способны образовывать водородные связи с молекулами воды, поэтому растворимы в ней. Низшие члены ряда смешиваются с водой в любых соотношениях, однако уже растворимость бутаналя в воде составляет 7%. Ароматические соединения в воде малорастворимы.
 Методы получения I. Получение альдегидов. 1. Реакции окисления алкенов озоном мольозонид (в реакции в зависимости от строения алкена образуются альдегиды и/или кетоны) Если озонолизу подвергается циклический алкен, образуется дикарбонильное соединение, например:
Методы получения I. Получение альдегидов. 1. Реакции окисления алкенов озоном мольозонид (в реакции в зависимости от строения алкена образуются альдегиды и/или кетоны) Если озонолизу подвергается циклический алкен, образуется дикарбонильное соединение, например:
 3. Прямое") 2. Окисление первичных спиртов в мягких условиях (см. тему «Свойства спиртов» ) 3. Прямое карбонилирование (оксосинтез) Этот метод получения альдегидов используется исключительно в промышленности. Алкены при обработке окисью углерода и водородом под давлением при повышенной температуре в присутствии октакарбонилдикобальта или других гомогенных металлокомплексных катализаторов образуют альдегиды. Решающий успех в оксосинтезе был достигнут благодаря использованию металлокомплексного катализа. Фактически это был первый пример гомогенного металлокомплексного катализа в органической химии, который сразу же приобрел важное значение для химической технологии.
2. Окисление первичных спиртов в мягких условиях (см. тему «Свойства спиртов» ) 3. Прямое карбонилирование (оксосинтез) Этот метод получения альдегидов используется исключительно в промышленности. Алкены при обработке окисью углерода и водородом под давлением при повышенной температуре в присутствии октакарбонилдикобальта или других гомогенных металлокомплексных катализаторов образуют альдегиды. Решающий успех в оксосинтезе был достигнут благодаря использованию металлокомплексного катализа. Фактически это был первый пример гомогенного металлокомплексного катализа в органической химии, который сразу же приобрел важное значение для химической технологии.
 4. Основным современным промышленным методом получения уксусного альдегида является каталитическое окисление этилена, получаемого в огромном количестве при крекинге углеводородов. Этилен окисляют в водном растворе, содержащем хлориды палладия (II) и меди (II) (так называемый «вакер-процесс» ). Катализатор регенерируют при действии кислорода в условиях непрерывного синтеза:
4. Основным современным промышленным методом получения уксусного альдегида является каталитическое окисление этилена, получаемого в огромном количестве при крекинге углеводородов. Этилен окисляют в водном растворе, содержащем хлориды палладия (II) и меди (II) (так называемый «вакер-процесс» ). Катализатор регенерируют при действии кислорода в условиях непрерывного синтеза:
 4. Восстановление производных карбоновых кислот Целый ряд функциональных производных карбоновых кислот - галогенангидриды, сложные эфиры, нитрилы, диалкиламиды могут быть в одну стадию превращены в альдегиды при действии специфических восстановителей. Например:
4. Восстановление производных карбоновых кислот Целый ряд функциональных производных карбоновых кислот - галогенангидриды, сложные эфиры, нитрилы, диалкиламиды могут быть в одну стадию превращены в альдегиды при действии специфических восстановителей. Например:
 II. Получение ароматических альдегидов. Многие реакции получения альдегидов ароматического ряда принципиально отличаются от методов синтеза алифатических альдегидов. Некоторые из них, основанные на реакции электрофильного ароматического замещения в бензольном кольце, были рассмотрены среди других процессов электрофильного ароматического замещения. 1. Окисление ароматических метилпроизводных. Метильная группа у ароматического кольца может быть окислена до альдегидной группы действием хромового ангидрида в растворе уксусного ангидрида. Выходы альдегидов редко превышают 50 -60%.
II. Получение ароматических альдегидов. Многие реакции получения альдегидов ароматического ряда принципиально отличаются от методов синтеза алифатических альдегидов. Некоторые из них, основанные на реакции электрофильного ароматического замещения в бензольном кольце, были рассмотрены среди других процессов электрофильного ароматического замещения. 1. Окисление ароматических метилпроизводных. Метильная группа у ароматического кольца может быть окислена до альдегидной группы действием хромового ангидрида в растворе уксусного ангидрида. Выходы альдегидов редко превышают 50 -60%.
 III. Получение кетонов. 1. Окисление вторичных спиртов 2. Гидратация алкинов по Кучерову
III. Получение кетонов. 1. Окисление вторичных спиртов 2. Гидратация алкинов по Кучерову
 3. Пиролиз сухих Са-, Ва-, Th- и других солей карбоновых кислот при 350 -450 о. С: Общим способом получения альдегидов и кетонов является щелочной гидролиз геминальных дигалогенопроизводных:
3. Пиролиз сухих Са-, Ва-, Th- и других солей карбоновых кислот при 350 -450 о. С: Общим способом получения альдегидов и кетонов является щелочной гидролиз геминальных дигалогенопроизводных:
 Химические свойства Реакционные центры: 1 – реакции по неподеленной электронной паре атома кислорода 2 – нуклеофильные реакции с участием частично положительно заряженного sp 2 -гиб-ридного атома углерода 3 – реакции по -углеродному атому 4 – реакции с участием связи С-Н 5 – реакции ароматических альдегидов и кетонов с участием бензольного кольца
Химические свойства Реакционные центры: 1 – реакции по неподеленной электронной паре атома кислорода 2 – нуклеофильные реакции с участием частично положительно заряженного sp 2 -гиб-ридного атома углерода 3 – реакции по -углеродному атому 4 – реакции с участием связи С-Н 5 – реакции ароматических альдегидов и кетонов с участием бензольного кольца
 Альдегиды и кетоны проявляют свойства слабых С-Н кислот.") 1. Кислотность (способность взаимодействовать с основаниями) Альдегиды и кетоны проявляют свойства слабых С-Н кислот. Появление частичного положительного заряда на атоме углерода карбонильной группы вызывает поляризацию связи С-Н и делает возможным отщепление протона, при этом устанавливается таутомерное кето-енольное равновесие, при нейтральном р. Н смещенное в сторону кетоформы: кето-форма енольная форма При обычных условиях содержание енольной формы невелико, например в ацетоне ее содержится 10 -4%.
1. Кислотность (способность взаимодействовать с основаниями) Альдегиды и кетоны проявляют свойства слабых С-Н кислот. Появление частичного положительного заряда на атоме углерода карбонильной группы вызывает поляризацию связи С-Н и делает возможным отщепление протона, при этом устанавливается таутомерное кето-енольное равновесие, при нейтральном р. Н смещенное в сторону кетоформы: кето-форма енольная форма При обычных условиях содержание енольной формы невелико, например в ацетоне ее содержится 10 -4%.
, у карбонильных соединений выражена слабо Карбонильные соединения") 2. Основность (способность взаимодействовать с протоном кислоты), у карбонильных соединений выражена слабо Карбонильные соединения относятся к числу очень слабых оснований, намного более слабых, чем вода и спирты. Так, например, ацетон оказывается наполовину протонированным только в 82%-й водной серной кислоте, т. е. в сильно кислом растворе.
2. Основность (способность взаимодействовать с протоном кислоты), у карбонильных соединений выражена слабо Карбонильные соединения относятся к числу очень слабых оснований, намного более слабых, чем вода и спирты. Так, например, ацетон оказывается наполовину протонированным только в 82%-й водной серной кислоте, т. е. в сильно кислом растворе.
 3. Реакции Н-замещения у -углеродного атома Активность С-Н связи в -положении молекулы повышена за счет электроноакцепторного действия (-I-эффект) карбонильной группы.
3. Реакции Н-замещения у -углеродного атома Активность С-Н связи в -положении молекулы повышена за счет электроноакцепторного действия (-I-эффект) карбонильной группы.
 Для присоединения к карбонильной группе ключевой стадией") 4. Реакции нуклеофильного присоединения (основная группа реакций) Для присоединения к карбонильной группе ключевой стадией является присоединение нуклеофила к электронодефицитному атому углерода. Нуклеофильный агент может быть или нейтральной молекулой (аммиак, первичные и вторичные амины, вода, спирты, тиолы, гидразин, гидроксиламин и др. ) или анионом (CN-, НSO 3 -; Н-, карбанионы и др. ). Присоединение нуклеофильных агентов по карбонильной группе является обратимым процессом. Исключение составляет реакция восстановления карбонильной группы с помощью комплексных гидридов Li. Al. Н 4; Na. BH 4; Li. BH 4, с образованием первичных или вторичных спиртов, а также присоединение металлоорганических соединений.
4. Реакции нуклеофильного присоединения (основная группа реакций) Для присоединения к карбонильной группе ключевой стадией является присоединение нуклеофила к электронодефицитному атому углерода. Нуклеофильный агент может быть или нейтральной молекулой (аммиак, первичные и вторичные амины, вода, спирты, тиолы, гидразин, гидроксиламин и др. ) или анионом (CN-, НSO 3 -; Н-, карбанионы и др. ). Присоединение нуклеофильных агентов по карбонильной группе является обратимым процессом. Исключение составляет реакция восстановления карбонильной группы с помощью комплексных гидридов Li. Al. Н 4; Na. BH 4; Li. BH 4, с образованием первичных или вторичных спиртов, а также присоединение металлоорганических соединений.
 Следует различать два типа обратимых реакций присоединения к карбонильной группе. В одном из них тетраэдрический анионный интермедиат протонируется по атому кислорода с образованием нейтрального конечного продукта присоединения. К такого рода процессам относятся образование ацеталей и кеталей, тиоацеталей и тиокеталей, циангидринов и присоединение енолят-ионов. В реакциях нуклеофильного присоединения другого типа нейтральный продукт присоединения подвергается дегидратации с образованием двойной связи между карбонильным углеродом и нуклеофильным агентом. Такой тип присоединения реализуется в реакциях карбонильных соединений с большой группой азотистых нуклеофильных реагентов: аммиака, первичных и вторичных аминов, гидроксиламина, семикарбазида, гидразина и его производных. Оба типа нуклеофильного присоединения могут быть выражены с помощью двух общих уравнений:
Следует различать два типа обратимых реакций присоединения к карбонильной группе. В одном из них тетраэдрический анионный интермедиат протонируется по атому кислорода с образованием нейтрального конечного продукта присоединения. К такого рода процессам относятся образование ацеталей и кеталей, тиоацеталей и тиокеталей, циангидринов и присоединение енолят-ионов. В реакциях нуклеофильного присоединения другого типа нейтральный продукт присоединения подвергается дегидратации с образованием двойной связи между карбонильным углеродом и нуклеофильным агентом. Такой тип присоединения реализуется в реакциях карбонильных соединений с большой группой азотистых нуклеофильных реагентов: аммиака, первичных и вторичных аминов, гидроксиламина, семикарбазида, гидразина и его производных. Оба типа нуклеофильного присоединения могут быть выражены с помощью двух общих уравнений:
 Альдегиды более реакционноспособны в реакциях присоединения нуклеофильных реагентов по сравнению с кетонами, и реакции присоединения к альдегидам характеризуются более высокими, чем для кетонов, значениями констант равновесия этого обратимого процесса.
Альдегиды более реакционноспособны в реакциях присоединения нуклеофильных реагентов по сравнению с кетонами, и реакции присоединения к альдегидам характеризуются более высокими, чем для кетонов, значениями констант равновесия этого обратимого процесса.
 а. Гидратация карбонильных соединений Равновесие сильно смещено влево в случае кетонов, формальдегид в воде на 99, 999% превращается в метандиол СН 2(ОН)2; уксусный альдегид гидратирован на 58%, для других альдегидов в водном растворе присутствуют в соизмеримом количестве исходное соединение и его гидратная форма.
а. Гидратация карбонильных соединений Равновесие сильно смещено влево в случае кетонов, формальдегид в воде на 99, 999% превращается в метандиол СН 2(ОН)2; уксусный альдегид гидратирован на 58%, для других альдегидов в водном растворе присутствуют в соизмеримом количестве исходное соединение и его гидратная форма.
 б. Присоединение спиртов с образованием ацеталей и кеталей. Продукт присоединения одного моля спирта к альдегиду или кетону называют соответственно полуацеталем или полукеталем: Ацетали и кетали образуются во второй стадии из полуацеталей или полукеталей присоединении второй молекулы спирта и дегидратации:
б. Присоединение спиртов с образованием ацеталей и кеталей. Продукт присоединения одного моля спирта к альдегиду или кетону называют соответственно полуацеталем или полукеталем: Ацетали и кетали образуются во второй стадии из полуацеталей или полукеталей присоединении второй молекулы спирта и дегидратации:
 в. Присоединение цианистого водорода Альдегиды и пространственно незатрудненные кетоны присоединяют цианистый водород с образованием циангидринов: Цианистоводородная (синильная) кислота является слабой кислотой с p. Kа=9, 2. Образующийся при ее депротонировании цианид-ион присоединяется к карбонильному углероду с образованием анионного интермедиата, протонирование которого приводит к циангидрину:
в. Присоединение цианистого водорода Альдегиды и пространственно незатрудненные кетоны присоединяют цианистый водород с образованием циангидринов: Цианистоводородная (синильная) кислота является слабой кислотой с p. Kа=9, 2. Образующийся при ее депротонировании цианид-ион присоединяется к карбонильному углероду с образованием анионного интермедиата, протонирование которого приводит к циангидрину:
 Медленной, определяющей скорость всей реакции стадией является присоединение цианид-иона к карбонильной группе.
Медленной, определяющей скорость всей реакции стадией является присоединение цианид-иона к карбонильной группе.
 г. Присоединение гидросульфита натрия Гидросульфит натрия присоединяется к карбонильной группе с образованием α-оксипроизводных сульфокислот: Гидросульфитные производные представляют собой кристаллические вещества. Исходные карбонильные соединения могут быть легко регенерированы при обработке гидросульфитных аддуктов кислотой или основанием - карбонатом натрия.
г. Присоединение гидросульфита натрия Гидросульфит натрия присоединяется к карбонильной группе с образованием α-оксипроизводных сульфокислот: Гидросульфитные производные представляют собой кристаллические вещества. Исходные карбонильные соединения могут быть легко регенерированы при обработке гидросульфитных аддуктов кислотой или основанием - карбонатом натрия.
 д. Реакции с металлорганическими соединениями. Синтез спиртов с помощью магнийорганических соединений.
д. Реакции с металлорганическими соединениями. Синтез спиртов с помощью магнийорганических соединений.
 е. Присоединение аммиака, первичных и вторичных аминов Классическим примером нуклеофильного присоединения по карбонильной группе являются реакции альдегидов и кетонов с первичными и вторичными аминами. Конечными продуктами реакции в случае аммиака и первичных аминов являются имины (основания Шиффа), а для вторичных аминов - енамины (енамин - ненасыщенный амин). В первой стадии реакции присоединения первичного амина образуется диполярный тетраэдрический интермедиат, который стабилизируется в результате переноса протона от азота к кислороду с образованием нейтрального полуаминаля (карбиноламина). Карбиноламин далее протонируется по атому кислорода. Отщепление воды от протонированной формы приводит к иминиевому катиону, который стабилизируется в результате отщепления протона в конечный продукт - альдимин или кетимин (основания Шиффа): 3
е. Присоединение аммиака, первичных и вторичных аминов Классическим примером нуклеофильного присоединения по карбонильной группе являются реакции альдегидов и кетонов с первичными и вторичными аминами. Конечными продуктами реакции в случае аммиака и первичных аминов являются имины (основания Шиффа), а для вторичных аминов - енамины (енамин - ненасыщенный амин). В первой стадии реакции присоединения первичного амина образуется диполярный тетраэдрический интермедиат, который стабилизируется в результате переноса протона от азота к кислороду с образованием нейтрального полуаминаля (карбиноламина). Карбиноламин далее протонируется по атому кислорода. Отщепление воды от протонированной формы приводит к иминиевому катиону, который стабилизируется в результате отщепления протона в конечный продукт - альдимин или кетимин (основания Шиффа): 3
 Соединения, образующиеся в результате взаимодействия альдегида или кетона с гидроксиламином, гидразином, фенилгидразином, 2, 4 -динитрофенилгидразином, nнитрофенилгидразином, семикарбазидом и тиосемикарбазидом, следует рассматривать как производные иминов. Большинство из них представляют собой кристаллические вещества, обладающие четкой температурой плавления. Эти производные используются для идентификации альдегидов и кетонов: гидроксиламин оксим
Соединения, образующиеся в результате взаимодействия альдегида или кетона с гидроксиламином, гидразином, фенилгидразином, 2, 4 -динитрофенилгидразином, nнитрофенилгидразином, семикарбазидом и тиосемикарбазидом, следует рассматривать как производные иминов. Большинство из них представляют собой кристаллические вещества, обладающие четкой температурой плавления. Эти производные используются для идентификации альдегидов и кетонов: гидроксиламин оксим
 гидразин 2, 4 -динитрофенилгидразин семикарбазид фенилгидразон семикарбазон
гидразин 2, 4 -динитрофенилгидразин семикарбазид фенилгидразон семикарбазон
 Общая схема и механизм Ad. N-реакции: Общая схема и механизм Ad. N-Е - реакции:
Общая схема и механизм Ad. N-реакции: Общая схема и механизм Ad. N-Е - реакции:
 Оксимы могут образовывать два вида пространственных изомеров: Е-изомер Z-изомер Е-оксимы в кислой среде подвергаются т. н. перегруппировке Бекмана, в результате которой получаются амиды карбоновых кислот: В этих условиях Z-оксимы дегидратируются до нитрилов.
Оксимы могут образовывать два вида пространственных изомеров: Е-изомер Z-изомер Е-оксимы в кислой среде подвергаются т. н. перегруппировке Бекмана, в результате которой получаются амиды карбоновых кислот: В этих условиях Z-оксимы дегидратируются до нитрилов.
 5. Восстановление Альдегиды и кетоны могут быть восстановлены до соответственно первичных или вторичных спиртов несколькими способами. Наиболее распространенным методом является восстановление комплексными гидридами: боргидридами натрия или лития и литийалюмогидридом.
5. Восстановление Альдегиды и кетоны могут быть восстановлены до соответственно первичных или вторичных спиртов несколькими способами. Наиболее распространенным методом является восстановление комплексными гидридами: боргидридами натрия или лития и литийалюмогидридом.
 Особое значение в органическом синтезе занимает восстановление карбонильной группы в ароматических и жирноароматических кетонах с помощью амальгамированного цинка и соляной кислоты по Э. Клемменсену (1913). Этот метод позволяет получать алкилбензолы с первичной алкильной группой, недоступные по реакции Фриделя-Крафтса:
Особое значение в органическом синтезе занимает восстановление карбонильной группы в ароматических и жирноароматических кетонах с помощью амальгамированного цинка и соляной кислоты по Э. Клемменсену (1913). Этот метод позволяет получать алкилбензолы с первичной алкильной группой, недоступные по реакции Фриделя-Крафтса:
 6. Окисление аммиачный раствор оксида серебра - реакция «серебряного зеркала» - качественная реакция на альдегиды.
6. Окисление аммиачный раствор оксида серебра - реакция «серебряного зеркала» - качественная реакция на альдегиды.
 Альдегиды легко окисляются до карбоновых кислот при действии самых разнообразных окислителей.
Альдегиды легко окисляются до карбоновых кислот при действии самых разнообразных окислителей.
 Кетоны окисляются до карбоновых кислот трудно и в жестких условиях (KMn. O 4, H 2 SO 4 конц. , t). Окисление кетонов возможно с разрывом углерод-углеродных связей и образованием смеси карбоновых кислот: (в общем случае 4 молекулы карбоновых кислот)
Кетоны окисляются до карбоновых кислот трудно и в жестких условиях (KMn. O 4, H 2 SO 4 конц. , t). Окисление кетонов возможно с разрывом углерод-углеродных связей и образованием смеси карбоновых кислот: (в общем случае 4 молекулы карбоновых кислот)
 Метилкетоны окисляются с помощью гипогалогенитов – галоформная реакция. Эта реакция используется не только для идентификации кетонов, но также часто оказывается полезной в синтезе, поскольку гипогалогенит не затрагивает двойную углерод – углеродную связь, например: Реакция протекает через стадии галогенирования и расщепления:
Метилкетоны окисляются с помощью гипогалогенитов – галоформная реакция. Эта реакция используется не только для идентификации кетонов, но также часто оказывается полезной в синтезе, поскольку гипогалогенит не затрагивает двойную углерод – углеродную связь, например: Реакция протекает через стадии галогенирования и расщепления:
 7. Присоединение галоген-нуклеофилов по карбонильной группе с последующим отщеплением + POCl 3
7. Присоединение галоген-нуклеофилов по карбонильной группе с последующим отщеплением + POCl 3
 поликонденсация фенола с формальдегидом линейный полимер При комнатной температуре образуется") 8. Реакции конденсации а) поликонденсация фенола с формальдегидом линейный полимер При комнатной температуре образуется преимущественно линейный полимер:
8. Реакции конденсации а) поликонденсация фенола с формальдегидом линейный полимер При комнатной температуре образуется преимущественно линейный полимер:
 При нагревании протекает конденсация с образованием разветвленных молекул:
При нагревании протекает конденсация с образованием разветвленных молекул:
 альдольная конденсация: Енолят-ионы подобно другим карбанионам (RC≡C-; CN-) способны обратимо присоединяться к карбонильной") б) альдольная конденсация: Енолят-ионы подобно другим карбанионам (RC≡C-; CN-) способны обратимо присоединяться к карбонильной группе альдегидов и кетонов. Эта реакция является общей для всех альдегидов, имеющих по крайней мере один атом водорода при α−углеродном атоме.
б) альдольная конденсация: Енолят-ионы подобно другим карбанионам (RC≡C-; CN-) способны обратимо присоединяться к карбонильной группе альдегидов и кетонов. Эта реакция является общей для всех альдегидов, имеющих по крайней мере один атом водорода при α−углеродном атоме.
 Механизм альдольной конденсации, катализируемой основанием, включает три стадии:
Механизм альдольной конденсации, катализируемой основанием, включает три стадии:
 В реакции участвуют две одинаковые или разные молекулы альдегидов или кетонов, С -атом одной из которых выступает как нуклеофил в отношении карбонильной группы другой молекулы. в) кротоновая конденсация:
В реакции участвуют две одинаковые или разные молекулы альдегидов или кетонов, С -атом одной из которых выступает как нуклеофил в отношении карбонильной группы другой молекулы. в) кротоновая конденсация:
 Перекрестная (смешанная) альдольная конденсация ароматических альдегидов с кетонами, приводящая к образованию α, β−ненасыщенных") г) Перекрестная (смешанная) альдольная конденсация ароматических альдегидов с кетонами, приводящая к образованию α, β−ненасыщенных жирноароматических кетонов, известна под названием реакции Кляйзена - Шмидта.
г) Перекрестная (смешанная) альдольная конденсация ароматических альдегидов с кетонами, приводящая к образованию α, β−ненасыщенных жирноароматических кетонов, известна под названием реакции Кляйзена - Шмидта.
 Сложноэфирная конденсация Кляйзена Сложноэфирная конденсация была открыта в 1887 году Л. Кляйзеном, который") д) Сложноэфирная конденсация Кляйзена Сложноэфирная конденсация была открыта в 1887 году Л. Кляйзеном, который при кипячении этилацетата с сухим этилатом натрия, не содержащим сольватного спирта, получил после подкисления реакционной смеси уксусной кислотой ацетоуксусный эфир с выходом 75%.
д) Сложноэфирная конденсация Кляйзена Сложноэфирная конденсация была открыта в 1887 году Л. Кляйзеном, который при кипячении этилацетата с сухим этилатом натрия, не содержащим сольватного спирта, получил после подкисления реакционной смеси уксусной кислотой ацетоуксусный эфир с выходом 75%.
 В реакции Перкина ароматические или гетероароматические альдегиды подвергаются конденсации в жестких условиях при") е) В реакции Перкина ароматические или гетероароматические альдегиды подвергаются конденсации в жестких условиях при 170 -200 °С с ангидридами алифатических кислот в присутствии солей карбоновых кислот в качестве основания.
е) В реакции Перкина ароматические или гетероароматические альдегиды подвергаются конденсации в жестких условиях при 170 -200 °С с ангидридами алифатических кислот в присутствии солей карбоновых кислот в качестве основания.
 бензоиновая конденсация Бензоиновая конденсация относится к числу самых старых реакций в органической химии,") ж) бензоиновая конденсация Бензоиновая конденсация относится к числу самых старых реакций в органической химии, которая была чисто случайно открыта еще в 1832 году Ю. Либихом и Ф. Велером при обработке бензальдегида цианидом калия. Образующий при этом α-гидроксикетон получил название бензоин, что дало название самой реакций конденденсации.
ж) бензоиновая конденсация Бензоиновая конденсация относится к числу самых старых реакций в органической химии, которая была чисто случайно открыта еще в 1832 году Ю. Либихом и Ф. Велером при обработке бензальдегида цианидом калия. Образующий при этом α-гидроксикетон получил название бензоин, что дало название самой реакций конденденсации.
 9. Полимеризация При стоянии альдегиды склонны к образованию циклических или полимерных ацеталей. Формальдегид при этом дает твердый линейный полимер, называемый параформальдегидом (параформ): Параформ при нагревании до 180 -200 о. С регенерируется в формальдегид. Формальдегид образует также циклический тример – триоксан, такой же триммер для ацетальдегида называется паральдегид: триоксан паральдегид
9. Полимеризация При стоянии альдегиды склонны к образованию циклических или полимерных ацеталей. Формальдегид при этом дает твердый линейный полимер, называемый параформальдегидом (параформ): Параформ при нагревании до 180 -200 о. С регенерируется в формальдегид. Формальдегид образует также циклический тример – триоксан, такой же триммер для ацетальдегида называется паральдегид: триоксан паральдегид
 Оба тримера при нагревании со следами кислот деполимеризуются до альдегидов. 37% водный раствор формальдегида называют формалином. В нем имеет место равновесие , которое сильно смещено вправо (К 103). В случае ацетона величина К, напротив, составляет всего 10 -2.
Оба тримера при нагревании со следами кислот деполимеризуются до альдегидов. 37% водный раствор формальдегида называют формалином. В нем имеет место равновесие , которое сильно смещено вправо (К 103). В случае ацетона величина К, напротив, составляет всего 10 -2.
 в отличие от насыщенных альдегидов, ароматические производные хлорируются по С-Н") 10. Специфические реакции. а) в отличие от насыщенных альдегидов, ароматические производные хлорируются по С-Н связи альдегидной группы:
10. Специфические реакции. а) в отличие от насыщенных альдегидов, ароматические производные хлорируются по С-Н связи альдегидной группы:
 реакция диспропорционирования (Канницаро) Окислительно - восстановительная реакция ароматических альдегидов была открыта в 1853") б) реакция диспропорционирования (Канницаро) Окислительно - восстановительная реакция ароматических альдегидов была открыта в 1853 г. выдающимся итальянским химиком С. Канниццаро. Альдегиды, не имеющие атома водорода при α-углеродном атоме, при нагревании в водноспиртовом растворе щелочи подвергаются диспропорционированию с образованием равных количеств первичного спирта и карбоновой кислоты. Эта реакция нашла широкое применение в ряду ароматических альдегидов. Реакция характерна для любых альдегидов, не имеющих атомов водорода у -углеродного атома. В противном случае в этих условиях протекает альдольная конденсация.
б) реакция диспропорционирования (Канницаро) Окислительно - восстановительная реакция ароматических альдегидов была открыта в 1853 г. выдающимся итальянским химиком С. Канниццаро. Альдегиды, не имеющие атома водорода при α-углеродном атоме, при нагревании в водноспиртовом растворе щелочи подвергаются диспропорционированию с образованием равных количеств первичного спирта и карбоновой кислоты. Эта реакция нашла широкое применение в ряду ароматических альдегидов. Реакция характерна для любых альдегидов, не имеющих атомов водорода у -углеродного атома. В противном случае в этих условиях протекает альдольная конденсация.