срс тубулопатия.pptx
- Количество слайдов: 40



Казахстанско Российский Медицинский Университет СРС на тему «Тубулопатии» Выполнила : Студентка 410 «Б» группы факультета ОМ Кулешова Анастасия Проверила:

ТУБУЛОПАТИЯ Группа нефропатий, в основе которых лежит патология транспортных систем вканальцах нефрона, характеризующаяся ранним частичным или генерализованным нарушением канальцевых функций при нормальной или несколько сниженной клубочковой фильтрации

Канальцевые функции • 1 - Реабсорбция ценных для организма неорганических и органических веществ, профильтровавшихся в клубочках; • 2 - Секреция в просвет канальцев веществ из крови и образующихся в клетках канальцев; • 3 - Концентрация мочи.

Проксимальные канальцы • Реабсорбция 60% профильтровавшегося натрия хлорида, калия, кальция и воды; • 80 -90% гидрокарбоната; • практически 100% - глюкозы и аминокислот; • Реабсорбция фосфатов контролируется паратгормоном; • Регуляция процесса «разведение – концентрация» мочи
 • Окончательная коррекция состава мочи по")
Дистальные канальцы – (извитой каналец и собирательные трубочки) • Окончательная коррекция состава мочи по содержанию натрия и хлоридов (основное место приложения альдостерона и вазопрессина); • Секреция калия; • Секреция водородных ионов (соединение с неабсорбируемыми анионами, в основном фосфатами, и аммиаком и выделение в виде титруемых кислот и аммония); • «Разведение – концентрация» мочи.

Классификация тубулопатий в зависимости от главного синдрома Наследственные тубулопатии, протекающие с полиурией • Наследственные тубулопатии, протекающие с деформацией скелета (рахитоподобные) • Наследственные тубулопатии, протекающие с нефролитиазом • • Первичные : Почечная глюкозурия, Почечный несахарный диабет, Почечный солевой диабет Вторичные: Нефронофтиз Фанкони, пиелонефрит, цистиноз, тирозинемия, ХПН Первичные: фосфат-диабет, болезнь де Тони-Дебре-Фанкони, почечный канальцевый ацидоз Вторичные (фенотипически сходные состояния): D-зависимый рахит, гипофосфатазия, целиакия, псевдогипопаратиреодизм Первичные: Цистинурия, глицинурия, имминоглицинурия, дистальный почечный тубулярный ацидоз Вторичные: оксалоз и вторичная гипероксалурия, ксантинурия, Синдром Леша-Нигана

Тубулопатии с ведущим синдромом полиурии

Клиника Почечная глюкозурия - Лаб. диагностика Генетика Потери сахара Слабость чувство голода Дегидратация и гипокалиемия. Экскреция глюкозы с мочой варьирует от 500 мг/сут до 100 г/сут, чаще 1 -30 г/сут без гипергликемии Аутосомно-доминантное наследование (тип А) Тип В – умеренная глюкозурия при синдроме глюкозо-галактозной мальабсорбции с кишечной дисфункцией (аутосомно-рецесивный тип) Почечный несахарный диабет - Полиурия Многократная рвота Судороги Дегидратация Гипонатриемия Полидипсия Повышение концетрации натрия, хлоридов, мочевины. Гипостенурия уд. вес мочи не более 1, 005 Х-сцепленная форма с мутацией в гене, кодирующем рецептор к АВП в клетках собирательных канальцев (V 2 R). Мутация гена аквапорина -2 Почечный солевой диабет - Ухудшение аппетита Рвота Потеря в весе Лихорадка Полиурия Гиперкалемия Низкий Na Высокий К Экскреция Na с мочой. Высокий ренин и альдостерон, задержка роста, дегидратация АР- Мутации в трех генах, кодирующих субъединицы эпителиального натриевого канала (ENAC) АД- Мутации гена, кодирующего минералокортикоидный

Почечная глюкозурия. Патогенез. Нарушение транспорта глюкозы при почечной гликозурии может быть связано с: • уменьшением анатомической массы проксимальных канальцев относительно гломерулярной поверхности • снижением функциональной способности системы, ответственной за транспорт глюкозы, против градиента концентрации в мембране канальцев клеток • снижением проницаемости для глюкозы клеточных мембран, обращённых в просвет канальцев; • снижением способности транспортировать глюкозу с помощью специфического мембранного носителя.

Почечная глюкозурия. Клиника. • Клинические проявления заболевания наблюдаются в очень тяжёлых случаях и обусловлены прежде всего значительными потерями сахара • Слабость, чувство голода, стойкий осмотический диурез • Дефицит углеводов приводит к задержке физического развития ребёнка и склонности к ацетонурии (особенно при голодании, лихорадочных состояниях). • Признаков почечной недостаточности не выявляется

Почечная глюкозурия. Диагностика. Диф. Диагностика. • Диагностируют наличием глюкозы в моче натощак и при нормальном уровне гликемии. • Подтверждают обнаружением глюкозы в не менее 3 порциях мочи и отсутствием изменений в гликемической кривой. • Наличие у пациента жажды, полиурии, гликозурии может навести на мысль о вероятности сахарного диабета. • Для исключения других тубулопатий, необходимо исследовать экскрецию фосфатов и аминокислот, показатели которых при изолированной почечной гликозурии не выходят за пределы нормы.

Почечная глюкозурия. Лечение. Прогноз. • Специального лечения в большинстве случаев не предусмотрено. • Рекомендуют сбалансированную диету. • Прогноз при почечной гликозурии принято считать благоприятным, так как, несмотря на постоянное выведение глюкозы с мочой, общее состояние пациентов обычно не нарушено

Почечный не сахарный диабет.
, ведущая к нарушению концентрирования")
Почечный несахарный диабет Нечувствительность дистальных канальцев к АДГ (антидиуретическому гормону), ведущая к нарушению концентрирования мочи, дегидратации и электролитным нарушениям (гипернатриемия, гиперхлоремия) Тип наследования: 90% Х-сцепленный, 10%-патология кодирования аквапоринового рецептора (А/Р, А/Д) Физиология: АДГ (аргинин-вазопрессин)=>вазопрессиновый рец. 2 типа собирательных трубочек=>стимуляция G-белка и активация аденилатциклазы=>повышается внутриклеточная ЦАМФ=>активация протеинкиназы. А и др. регуляторных белков=>увеличение проницаемости клеточной мембраны для воды
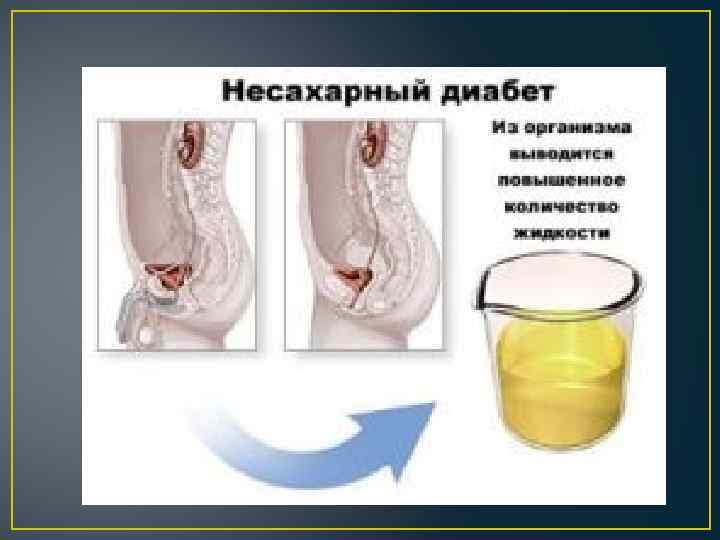


Почечный несахарный диабет. Клиника. • • • Ранний манифест При грудном вскармливании – отсроченный Обезвоживание Интермиттирующая лихорадка Стойкие запоры Полиурия (до 8 -18 л/сут), никтурия, энурез Мегацистис, мегауретер, гидронефроз Нарушение роста Задержка психического развития (олигофрения) Внутримозговой кальциноз

Почечный несахарный диабет. Диагностика. Проба с вазопрессином(интраназально детям до года - 10 мкг, старшего возраста - 20 мкг) - оценка через 2 часа осмолярности мочи (норма - выше 800 м. Осм/кг воды). При осмолярности ниже 200 м. Осм/кг - диагностируют почечную форму несахарного диабета. Уровень АДГ в крови - норма.

Лечение: • Обеспечение достаточным количеством жидкости; • Ограничение в диете белка до 2 г/кг на короткое время, натрия до 1 ммоль/кг. • Тиазидовые диуретики - гидрохлортиазид 2 мг/кг/сут (снижается натрий-зависимая реабсорбции хлора в дистальных канальцах); • Калийсберегающие диуретики - амилорид 10 -20 мг/кв. м/сут у детей после 6 лет (блокирует каналы реабсорбции натрия в собирательных трубочках, снижает внеклеточное содержание натрия в интерстициальной ткани, уменыпает объем внеклеточной жидкости, снижает реабсорбцию натрия в проксимальных каналыдах); • НПВС - индометацин 2 мг/кг/сут (усиливает действие гипотиазида).
")
Почечный солевой диабет (Псевдогипоальдостеронизм)

Псевдогипоальдестеронизм 1 тип – первичный, почечный • А/Д тип наследования Патофизиология: дефект минералокортикоидного рецептора 1 типа. Первично-нарушена реабсорбция Na в канальцах, вторично - дефект K-Na-АТФазы Клиника: • в/у – гидроамнион; • с 1 недели – обильное срыгивание, дегидратация, потеря массы; • нефрокальциноз на фоне гипер-Са-урии; • Парциальные формы яркой клиники не имеют. Б/х: Гипо-Na-еми, гипер-K-емия, метабол. ацидоз, гипер-Na-урия Гормоны: норма или повышение ренина и альдостерона крови Биопсия: гиперплазия ЮГА (не всегда) Терапия: заместительная – 3 -6 г/сут Na. Cl Прогноз: относительно благополучный. Чувствительность к минералокортикоидам повышается после 1 -2 лет жизни.

Псевдогипоальдестеронизм 1 тип – первичный, полиорганный • Нечувствительность к минералокортикоидам клеток-мишеней (почки, толстая кишка, потовые и слюнные железы) • А/Р тип наследования (рецепторы 1 типа не поражаются, изменен амилоид-чувствительный эпителиальный канал реабсорбции Na, как при синдроме Лиддла) Клиника: тяжелое мочеизнурение с выраженной дегидратацией Лечение: • Диета с ограничением К; • Ионообменные смолы; • Индометацин (? ); • Заместительная терапия Na. Cl малоэффективна • Минералокортикоиды не оказывают воздействия Прогноз: тяжелый
 • Наследственная а/д тубулопатия (гиперальдостеронизм и минимальная секреция альдостерона) Клиника: •")
ПСЕВДОАЛЬДОСТЕРОНИЗМ (синдром Лиддла) • Наследственная а/д тубулопатия (гиперальдостеронизм и минимальная секреция альдостерона) Клиника: • полиурия, полидипсия, • задержка психомоторного развития, • гипокалиемия, алкалоз, артериальная гипертензия. Диагностика: низкий уровень альдостерона, отсутствие ренина в сыворотке. Патофизиология: активация реабсорбции ионов натрия через эпителиальные натриевые каналы (в различных органах) ведет к артериальной гипертензии и снижению реабсорбции ионов калия. Лечение - коррекция гипокалиемии (триамтерен - 10

Тубулопатии с ведущим синдромом нефролитиаза.
 - Генетика Отставание в росте")
Клиника Синдром Баттлера. Олбрайта (дистальн ый тубулярны й ацидоз) - Генетика Отставание в росте Кризы обезвоживани Полиурия Нефрокальциноз Мочекаменная болезнь Тугоухость Признаки остеопороза. Кальцификаты в мозговом слое почек. Нефролитиаз. Выраженный системный ацидоз, увеличение р. Н мочи, гипо. К, гипер. Са. Диагностика заключается в использовании теста с хлоридом аммония или хлоридом кальция - р. Н мочи не ниже 6, 0. Тип наследования: Аутосомнорецессивный, аутосомнодоминантный или спорадические случаи. Начало клинических проявлений с 10 -20 лет Боли в животе Приступы почечной колики Нарушения уродинамики АГ Кристаллы цистина в моче при микроскопии Выявление аминоацидурии при хроматографии мочи Наследуется по аутосомнорециссивному типу. Все типы цистинурии развиваются при мутациях гена SLC 3 A 1. Частота встречаемости — 1: 20000. Выявление увеличенной суточной экскреции глицина с мочой (более 1 г в сут) при нормальном содержании глицина в плазме и отсутствии нарушений реабсорбции других аминокислот. Аутосомнодоминантн ы. Й тип наследования. Цистинури я - Глицинурия - Нефролитиаз - Пиелонефрит - Возможное развитие ХПН - Лаб. Диагностика
")
Синдром Баттлера-Олбрайта (дистальный тубулярный ацидоз)
. Б/х маркеры. Метаболический ацидоз Умеренная гипофосфатемия Гипокальциемия Повышение уровня")
Синдром Баттлера-Олбрайта (дистальный тубулярный ацидоз). Б/х маркеры. Метаболический ацидоз Умеренная гипофосфатемия Гипокальциемия Повышение уровня щелочной фосфатазы крови Снижение экскреции титруемых кислот и аммиака Щелочная или нейтральная РН Гипостенурия

Диагностика • Метаболический гиперхлоремический ацидоз с нормальными показателями анионной разницы плазмы • ( Na-Cl-HCO 3 = 8 -16 ммольл) • Щелочная реакция мочи (р. Н >5, 5 при выраженном ацидозе или при нагрузке хлоридом аммония 0, 1 гкг) • Нефрокальциноз • Положительная анионная разница мочи ( Na+K>Cl) • Нормальная фракционная экскреция бикарбоната ( не более 5%) при восстановлении нормального уровня бикарбоната сыворотки • Низкая разница между р. СО 2 щелочной мочи и р. СО 2 сыворотки крови (не более 20 мм рт ст)

Клиника • Заболевание проявляется после 2 -лет, иногда начало может быть прослежено с первых месяцев жизни в виде тошноты, запоров, анорексии, полиурии, дегидратации, отставания в развитии. • Характерна задержка в росте, часто наблюдается рахит, остеомаляция, боли в костях, патологические переломы, периодические мышечные параличи. • У больных могут быть внезапные кризы дегидратации, сочетающиеся с расстройством кровообращения, сердечной аритмией, тошнотой, вялыми параличами, затрудненным дыханием, сонливостью, комой. • Постоянно щелочная моча, ее р. Н не понижается до минимального уровня (6, 0), несмотря на системный ацидоз (р. Н крови ниже 7, 3; SВ ниже 20 мэкв/л). Экскреция титруемых кислот и аммония снижены в соответствии с повышенным р. Н мочи.

Лечение. Прогноз. • Лечение должно быть направлено на восстановление роста, ликвидацию изменений в костях, профилактику дальнейшего отложения кальция в почках, что осуществляется с помощью ощелачивающей терапии в виде раствора Олбрайта, состоящего из 98 г цитрата натрия, 140 г лимонной кислоты, воды до 1 л. • При подборе индивидуальной дозы ориентируются на уровень р. Н и стандартных бикарбонатов крови. При гипокалиемии к раствору добавляют цитрат калия. • Прогноз благоприятен при рано начатом (до появления нефрокальциноза) лечении. В случае присоединения нефрокальциноза часто развивается интерстициальный нефрит и заболевание прогрессирует до стадии ХПН.

Цистинурия.

Патогенез В основе заболевания лежит нарушение транспорта цистина в слизистой оболочке тонкой кишки и в почечных канальцах. Есть 3 типа цистинурии: • 1 тип — отсутствие транспорта цистина и диаминомонокарбоновых аминокислот в кишечнике. • 2 тип — снижение до 50 % транспорта цистина в почках и полное отсутствие транспорта диаминомонокарбоновых аминокислот в кишечнике и почках. • 3 тип — снижение транспорта этих аминокислот в почках при нормальном их всасывании в кишечнике.

Клиника • Чаще всего начало клинических проявлений с 10 -20 летнего возраста. Проявлениями могут быть боли в животе, приступы почечной колики, нарушения уродинамики, артериальная гипертензия, отставание в физическом развитии. В анализах: кристаллы цистина в моче при микроскопии, выявление аминоацидурии при хроматографии мочи. Диагноз и дифдиагноз Диагноз выставляется только при количественном определении цистина в крови и моче. Дифференцируют цистинурию с пиелонефритом, интерстициальным нефритом, нефролитиазом, аминоацидурией, ХПН. Лечение • Режим активный. Диета картофельная, с ограничением серусодержащих белков, ограничение в пище метионина. Увеличение питьевого режима. Щелочное питье (р. Н мочи необходимо поддерживать на уровне 7, 5).

Глицинурия.

Глицинурия. • одна из форм нарушения обмена веществ, сопровождающаяся повышенной экскрецией глицина с мочой (более 200 миллиграмм в сутки). Следствием Глицинурия могут быть вторичные изменения почек и мочевыводящих путей. • Глицинурия, обусловленная нарушением реабсорбции глицина в почечных канальцах, характеризуется массивной экскрецией глицина (до 600 миллиграмм в сутки). Одним из проявлений этой формы Глицинурия является нефролитиаз

• Глюкоглицинурия наряду с повышенным выделением глицина с мочой характеризуется клиникой сахарного диабета. • Специфические методы лечения наследственно обусловленных первичных Глицинурия не разработаны. • Прогноз неблагоприятен при развитии уремии. Тяжесть заболевания определяется степенью поражения почек, центральная нервная система и островков поджелудочной железы. • Глицинурия как симптом отмечается при различных нарушениях транспорта аминокислот в проксимальных почечных канальцах. Она может быть наследственно обусловленной (например, при синдроме Лoy, синдроме де Тони — Дебре — Фанкони и другие) и приобретённой (при диффузном гломерулонефрите, пиелонефрите, медикаментозных поражениях канальцев почек и тому подобное). • Лечение сводится к терапии основного заболевания.

Литература. • Игнатова М. С. и Вельтищев Ю. Е. Детская нефрология, с. 257. Л. , 1989. • Шилов Е. М Нефрология. С. 688 2007. • Цигин Нефрология. С. 389 2010.
срс тубулопатия.pptx