

Prezentatsia_immunodifitsity_29_10_2012.ppt
- Количество слайдов: 43
 Авторы, год Дж.") История исследования HLA Открытия Первый антиген гистосовместимости человека Mac (HLA-A 2) Авторы, год Дж. Дассэ, 1958 г Доказана ведущая роль HLA антигенов в развитии реакции Дж. ван Рууд и др, 1966 г отторжения трансплантата Установлена корреляция между аллельными вариантами З. Фалчук и др. , 1972 г HLA антигенов и определенными заболеваниями Установлена структура HLA антигенов класса I К. Накамура и др, 1973 г Показана роль антигенов гистосовместимости в ограничении иммунного ответа (двойное распознавание) Определена и выделена аминокислотная последовательность антигенов HLA класса II Продемонстрирован биохимический полиморфизм HLA антигенов Определена пространственная структура HLA-A 2 антигена Р. Цинкернагель, П. Доэрти, 1974 г Г. Кратцин и др. , 1981 г. Р. Василов и др. , 1983 г П. Бeркман и др. 1987 г. Установлен характер распределения HLA антигенов в 1991 – 1993 гг. большинстве этнических групп планеты
История исследования HLA Открытия Первый антиген гистосовместимости человека Mac (HLA-A 2) Авторы, год Дж. Дассэ, 1958 г Доказана ведущая роль HLA антигенов в развитии реакции Дж. ван Рууд и др, 1966 г отторжения трансплантата Установлена корреляция между аллельными вариантами З. Фалчук и др. , 1972 г HLA антигенов и определенными заболеваниями Установлена структура HLA антигенов класса I К. Накамура и др, 1973 г Показана роль антигенов гистосовместимости в ограничении иммунного ответа (двойное распознавание) Определена и выделена аминокислотная последовательность антигенов HLA класса II Продемонстрирован биохимический полиморфизм HLA антигенов Определена пространственная структура HLA-A 2 антигена Р. Цинкернагель, П. Доэрти, 1974 г Г. Кратцин и др. , 1981 г. Р. Василов и др. , 1983 г П. Бeркман и др. 1987 г. Установлен характер распределения HLA антигенов в 1991 – 1993 гг. большинстве этнических групп планеты
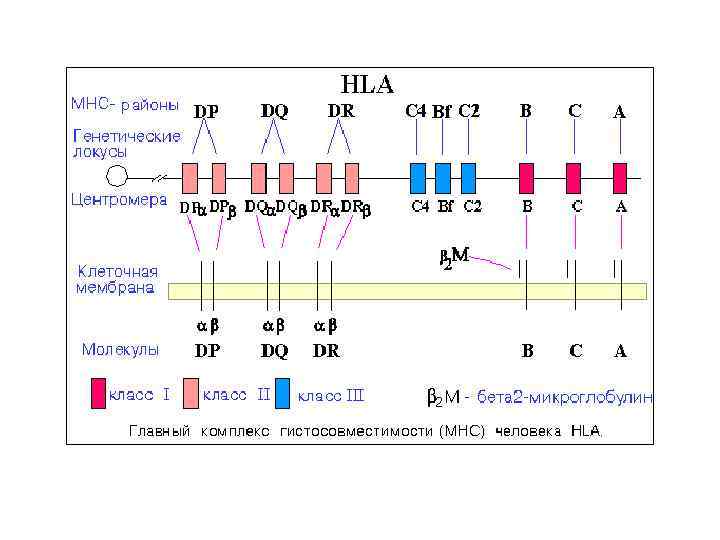
 Структура HLA локуса • MHC регион является одним из наиболее сложных областей генома для изучения. • Этот регион составляет ~ 4 МБ нуклеотидов и содержит более 160 генов. • Эти гены кодируют примерно 40 процентов белков, участвующих в иммунной системе, включая лейкоцитарные антигены, мембранные гликопротеины, которые являются посредниками сигнализации Т-лимфоцитов. • MHC регион широко изучен и предлагается, что он определяет большинство воспалительных и аутоиммунных расстройств
Структура HLA локуса • MHC регион является одним из наиболее сложных областей генома для изучения. • Этот регион составляет ~ 4 МБ нуклеотидов и содержит более 160 генов. • Эти гены кодируют примерно 40 процентов белков, участвующих в иммунной системе, включая лейкоцитарные антигены, мембранные гликопротеины, которые являются посредниками сигнализации Т-лимфоцитов. • MHC регион широко изучен и предлагается, что он определяет большинство воспалительных и аутоиммунных расстройств
 Антигены гистосовместимости • • HLA-комплекс – антигены HLA-комплекса (HLA – human leukocyte antigen – человеческий антиген лейкоцитов) – антигены гистосовместимости (то есть, генетически детерминированные изоантигены, которые вызывают иммунный ответ при трансплантации в организм другого человека) - главный комплекс гистосовместимости (MHC) Гены MHC – находится на коротком плече 6 хромосомы. Молекулы I класса – HLA-A, HLA-B и HLA-C – кодируются тремя отдельными парами генных локусов. Антигены I класса, впервые найденные на лейкоцитах (отсюда термин HLA), экспрессируются (синтезируются и выводятся на клеточную поверхность) почти во всех тканях (продукт четвертого локуса I класса, HLA-G, экспрессируется только в трофобласте). Молекулы I класса играют важную роль при распознавании антигена цитотоксическими T-клетками (CD 8). • Молекулы II класса кодируются тремя или более генными локусами (DR, DP и DQ). HLA-DR антигены известны также как Ia антигены по аналогии с антигенами иммунного ответа у мышей. Антигены II класса имеют ограниченное распространение в тканях, преимущественно на B-клетках, макрофагах, обрабатывающих антиген, и активированных T-клетках; они участвуют в распознавании антигена T-клетками (хелперами; CD 4). • В 6 хромосоме между генами I и II классов находятся гены, кодирующие молекулы III класса (которые включают факторы комплемента 2, 4 a и 4 b) и цитокины TNFα и TNFβ.
Антигены гистосовместимости • • HLA-комплекс – антигены HLA-комплекса (HLA – human leukocyte antigen – человеческий антиген лейкоцитов) – антигены гистосовместимости (то есть, генетически детерминированные изоантигены, которые вызывают иммунный ответ при трансплантации в организм другого человека) - главный комплекс гистосовместимости (MHC) Гены MHC – находится на коротком плече 6 хромосомы. Молекулы I класса – HLA-A, HLA-B и HLA-C – кодируются тремя отдельными парами генных локусов. Антигены I класса, впервые найденные на лейкоцитах (отсюда термин HLA), экспрессируются (синтезируются и выводятся на клеточную поверхность) почти во всех тканях (продукт четвертого локуса I класса, HLA-G, экспрессируется только в трофобласте). Молекулы I класса играют важную роль при распознавании антигена цитотоксическими T-клетками (CD 8). • Молекулы II класса кодируются тремя или более генными локусами (DR, DP и DQ). HLA-DR антигены известны также как Ia антигены по аналогии с антигенами иммунного ответа у мышей. Антигены II класса имеют ограниченное распространение в тканях, преимущественно на B-клетках, макрофагах, обрабатывающих антиген, и активированных T-клетках; они участвуют в распознавании антигена T-клетками (хелперами; CD 4). • В 6 хромосоме между генами I и II классов находятся гены, кодирующие молекулы III класса (которые включают факторы комплемента 2, 4 a и 4 b) и цитокины TNFα и TNFβ.
 Динамика открытия новых антигенов/аллелей с 1968 по 2002 годы
Динамика открытия новых антигенов/аллелей с 1968 по 2002 годы
 АЛЛЕЛЬНЫЕ ВАРИАНТЫ HLA ЛОКУСА
АЛЛЕЛЬНЫЕ ВАРИАНТЫ HLA ЛОКУСА
 АЛЛЕЛЬНЫЕ ВАРИАНТЫ HLA ЛОКУСА
АЛЛЕЛЬНЫЕ ВАРИАНТЫ HLA ЛОКУСА
 АЛЛЕЛЬНЫЕ ВАРИАНТЫ HLA ЛОКУСА HLA Class II - DRB Alleles Gene Alleles DRB 1 DRB 2 DRB 3 DRB 4 DRB 5 DRB 6 DRB 7 DRB 8 DRB 9 1, 051 1 57 15 19 3 2 1 1 Proteins 792 0 46 8 16 0 0 Nulls 15 0 0 3 2 0 0
АЛЛЕЛЬНЫЕ ВАРИАНТЫ HLA ЛОКУСА HLA Class II - DRB Alleles Gene Alleles DRB 1 DRB 2 DRB 3 DRB 4 DRB 5 DRB 6 DRB 7 DRB 8 DRB 9 1, 051 1 57 15 19 3 2 1 1 Proteins 792 0 46 8 16 0 0 Nulls 15 0 0 3 2 0 0
 АЛЛЕЛЬНЫЕ ВАРИАНТЫ HLA ЛОКУСА HLA Class II Gene DRA DRB DQA 1 DQB 1 DPA 1 DPB 1 DMA DMB DOA DOB Alleles 7 1, 150 46 160 33 150 7 13 12 13 Proteins 2 862 29 111 16 130 4 7 3 5 Nulls 0 20 1 1 0 3 0 0 1 0
АЛЛЕЛЬНЫЕ ВАРИАНТЫ HLA ЛОКУСА HLA Class II Gene DRA DRB DQA 1 DQB 1 DPA 1 DPB 1 DMA DMB DOA DOB Alleles 7 1, 150 46 160 33 150 7 13 12 13 Proteins 2 862 29 111 16 130 4 7 3 5 Nulls 0 20 1 1 0 3 0 0 1 0
 Аллельный полиморфизм некоторых генов системы HLA класса II. Гены системы HLA DRA Количество известных аллелей 2 DRB 1 316 DRB 2 1 DRB 3 38 DRB 4 12 DRB 5 17 DRB 6 3 DRB 7 2 DRB 8 1 DRB 9 1 DQA 1 23 DQB 1 54 DPA 1 21 DPB 1 101
Аллельный полиморфизм некоторых генов системы HLA класса II. Гены системы HLA DRA Количество известных аллелей 2 DRB 1 316 DRB 2 1 DRB 3 38 DRB 4 12 DRB 5 17 DRB 6 3 DRB 7 2 DRB 8 1 DRB 9 1 DQA 1 23 DQB 1 54 DPA 1 21 DPB 1 101

 Механизмы развития аутоиммунных заболеваний Механизмы Контакт с иммунной системой скрытых антигенов Антигены, вовлеченные в патогенез Тиреоглобулин (? ) Причины развития Аутоиммунные заболевания В норме тиреоглобулин скрыт в фолликулах щитовидной железы Белки хрусталика Хрусталик не имеет сосудов, в норме белки скрыты от иммунной системы Антигены возникают в сперматозоидов постнатальной жизни Лекарства, вирусные Присоединение гаптенов, частичное и другие инфекции разрушение Тиреоидит Хашимото Симпатический офтальмит Бесплодие (у мужчин) ревматические болезни Снижение концентрации супрессорных антител Снижение количества Тсупрессоров Активация супрессированных клонов лимфоцитов Многие типы Появление "запрещенных" клонов Опухолевая трансформация лимфоцитов; злокачественная лимфома и лимфоцитарная лейкемия Антистрептококковые Антитела против внешних антигенов антитела и действуют на собственные антигены миокардиальные антигены Различные типы Потеря контроля над иммунным ответом в результате недостатка Ir Повреждение собственных антигенов Перекрестный иммунитет на внешние и собственные антигены Нарушения в генах иммунного ответа (Ir антигенах) Многие типы Вирус Эпштейн. Барра; ? другие вирусы Многие типы Дефицит В-клеток; врожденная агаммаглобулинемия Брутона Дефицит Т-клеток, поствирусные инфекции Стимуляция В-клеток Редко наблюдается Ревматоидный артрит Гемолитическая анемия, тромбоцитопения Ревматические заболевания Многие типы1
Механизмы развития аутоиммунных заболеваний Механизмы Контакт с иммунной системой скрытых антигенов Антигены, вовлеченные в патогенез Тиреоглобулин (? ) Причины развития Аутоиммунные заболевания В норме тиреоглобулин скрыт в фолликулах щитовидной железы Белки хрусталика Хрусталик не имеет сосудов, в норме белки скрыты от иммунной системы Антигены возникают в сперматозоидов постнатальной жизни Лекарства, вирусные Присоединение гаптенов, частичное и другие инфекции разрушение Тиреоидит Хашимото Симпатический офтальмит Бесплодие (у мужчин) ревматические болезни Снижение концентрации супрессорных антител Снижение количества Тсупрессоров Активация супрессированных клонов лимфоцитов Многие типы Появление "запрещенных" клонов Опухолевая трансформация лимфоцитов; злокачественная лимфома и лимфоцитарная лейкемия Антистрептококковые Антитела против внешних антигенов антитела и действуют на собственные антигены миокардиальные антигены Различные типы Потеря контроля над иммунным ответом в результате недостатка Ir Повреждение собственных антигенов Перекрестный иммунитет на внешние и собственные антигены Нарушения в генах иммунного ответа (Ir антигенах) Многие типы Вирус Эпштейн. Барра; ? другие вирусы Многие типы Дефицит В-клеток; врожденная агаммаглобулинемия Брутона Дефицит Т-клеток, поствирусные инфекции Стимуляция В-клеток Редко наблюдается Ревматоидный артрит Гемолитическая анемия, тромбоцитопения Ревматические заболевания Многие типы1
 DR 5") Ассоциация HLA с заболеваниями Заболевание Аллель HLA ОР Аутоиммунный тиреодит (болезнь Хасимото) DR 5 3, 2 Ревматоидный артрит DR 4 5, 8 Герпетиформный дерматит DR 3 56, 4 Хронический гепатит (аутоиммунный) DR 3 13, 9 Кишечный инфатилизм DR 3 10, 8 Синдром Шегрена DR 3 9, 7 Болезнь Аддисона DR 3 6, 3 Инсулинозависимый диабет DR 3; DR 4 5, 0; 6, 8 Тиреотоксикоз DR 3 3, 7 Первичная микседема DR 3 5, 7 Синдром Гудпасчера DR 2 13, 1 Туберкулезная лепра DR 2 8, 1 Рассеянный склероз DR 2 4, 8 а. Ассоциированные с классом II
Ассоциация HLA с заболеваниями Заболевание Аллель HLA ОР Аутоиммунный тиреодит (болезнь Хасимото) DR 5 3, 2 Ревматоидный артрит DR 4 5, 8 Герпетиформный дерматит DR 3 56, 4 Хронический гепатит (аутоиммунный) DR 3 13, 9 Кишечный инфатилизм DR 3 10, 8 Синдром Шегрена DR 3 9, 7 Болезнь Аддисона DR 3 6, 3 Инсулинозависимый диабет DR 3; DR 4 5, 0; 6, 8 Тиреотоксикоз DR 3 3, 7 Первичная микседема DR 3 5, 7 Синдром Гудпасчера DR 2 13, 1 Туберкулезная лепра DR 2 8, 1 Рассеянный склероз DR 2 4, 8 а. Ассоциированные с классом II
 Ассоциация HLA с заболеваниями б. Ассоциированные с классом I, HLA-B 27 Анкилозирующий спондилоартрит B 27 87, 4 Болезнь Рейтера B 27 37, 0 Сальмонелезный артрит B 27 29, 7 Шигеллезный артрит B 27 20, 7 Ерсиниозный артрит B 27 17, 6 Гонококковый артрит B 27 14, 0 Увеит B 27 14, 6 Амилоидоз при ревматоидном артрите B 27 8, 2 Подострый тиреодит Bw 35 13, 7 Псориаз Cw 6 13, 3 Спонтанный гемохроматоз A 3 8, 2 Миастения B 8 4, 4 в. Другие болезни, ассоциированные с классом I
Ассоциация HLA с заболеваниями б. Ассоциированные с классом I, HLA-B 27 Анкилозирующий спондилоартрит B 27 87, 4 Болезнь Рейтера B 27 37, 0 Сальмонелезный артрит B 27 29, 7 Шигеллезный артрит B 27 20, 7 Ерсиниозный артрит B 27 17, 6 Гонококковый артрит B 27 14, 0 Увеит B 27 14, 6 Амилоидоз при ревматоидном артрите B 27 8, 2 Подострый тиреодит Bw 35 13, 7 Псориаз Cw 6 13, 3 Спонтанный гемохроматоз A 3 8, 2 Миастения B 8 4, 4 в. Другие болезни, ассоциированные с классом I

 Строение иммуноглобулина Ig. G •
Строение иммуноглобулина Ig. G •
 Схема клеточного взаимодействия при гуморальном иммунитете Примечание: АГ - антиген, CD 4, 8 Т - "ранние" Т-хелперы, CD 4 Т - "поздние" Т-хелперы, В-клетки, ПК - плазматические клетки
Схема клеточного взаимодействия при гуморальном иммунитете Примечание: АГ - антиген, CD 4, 8 Т - "ранние" Т-хелперы, CD 4 Т - "поздние" Т-хелперы, В-клетки, ПК - плазматические клетки
 Распознавание антигена Т-лимфоцитами
Распознавание антигена Т-лимфоцитами
 Хромосомная локализация генов, кодирующих иммунноглобулины и антигенсвязывающие рецепторы человека Пептид Ig. H (тяжелые цепи иммуноглобулина) Хромосома человека 14, 15, 16 λ- цепь (легкие цепи иммунноглобулинов) 22 κ - цепь (легкие цепи ммунноглобулинов) α - цепь Тк. Р 2 14 β - цепь Тк. Р 7 γ - цепь Тк. Р 7 δ - цепь Тк. Р 14 β 2 – микроглобулин 15
Хромосомная локализация генов, кодирующих иммунноглобулины и антигенсвязывающие рецепторы человека Пептид Ig. H (тяжелые цепи иммуноглобулина) Хромосома человека 14, 15, 16 λ- цепь (легкие цепи иммунноглобулинов) 22 κ - цепь (легкие цепи ммунноглобулинов) α - цепь Тк. Р 2 14 β - цепь Тк. Р 7 γ - цепь Тк. Р 7 δ - цепь Тк. Р 14 β 2 – микроглобулин 15


 гена (генов), локус. OMIM – Online") Наследственные болезни иммунитета Указаны: код ОMIM, имя (имена) гена (генов), локус. OMIM – Online Mendelian Inheritance in Man (Менделевское наследование человека в Сети) 100000 – аутосомно-диминантный тип наследования (информация до 15 мая 1994 года), 200000 - аутосомно-рецессивное наследование, 300000 - локусы и фенотипы, связанные с X-хромосомой, 400000 - локусы и фенотипы, связанные с Y-хромосомой, 500000 – локусы и фенотипы, связанные с митохондральным (цитоплазматическим) наследованием, 600000 – аутосомное наследование (информация после 15 мая 1994 г). Агаммаглобулинемия сцепленная с хромосомой X (брутоновская), 300300, ВТК, AGMX 1, IMD 1, XLA, AT, Xq 21. 3 -q 22 Атаксия-телеангиэктазия, 208900, ATM, ATA, ATI, 208900, IIq 22. 3 Болезнь Коудена, 158350, PTEN, MMAC 1, 601728, 10 q 23. 3 Гипериммуноглобулинемия, 144120, Gl, IGHR, 14 q 32. 33 Гломерулонефрит мембранозно пролиферативный, 134370 (фактор Н) HF 1, CFH, HUS, , Iq 32 Дефекты CD: • 186780, CD 3 Z (S-субъединица рецептора Т-лимфоцитов), CD 3 Z, TCRZ, Iq 22 -q 23 • 186940, CD 4+, CD 4, 186940, 12 pter-pl 2 • 107271, CD 59, 11 р13 • 146760, CD 64, FCGR 1 A, IGFR 1, CD 64, Iq 21. 2 -q 21. 3 Дефект адгезии лейкоцитов, 116920, ITGB 2 (интегрин), CD 18, LCAMB, LAD, 600065, 21 q 22. 3 Дефект иммунного интерферона, 147570, IFNG, 12 ql 4 Дефект а-интерферона, 147660, IFNA 1, 9 р22 Дефект лёгких цепей Ig к-Ig, 147200, IGKC, 2 р12 Дефект а-цепи рецептора интерлейкина-2, 147730, IL 2 RA, IL 2 R, 10 р15 -р14 Дефицит инактиватора компонента комплемента 217030, СЗЬ, IF, 4 q 25 Дефицит лактоферрина нейтрофилов, 245480, LTF, 150210 (лактоферрин), 3 q 21 -q 23
Наследственные болезни иммунитета Указаны: код ОMIM, имя (имена) гена (генов), локус. OMIM – Online Mendelian Inheritance in Man (Менделевское наследование человека в Сети) 100000 – аутосомно-диминантный тип наследования (информация до 15 мая 1994 года), 200000 - аутосомно-рецессивное наследование, 300000 - локусы и фенотипы, связанные с X-хромосомой, 400000 - локусы и фенотипы, связанные с Y-хромосомой, 500000 – локусы и фенотипы, связанные с митохондральным (цитоплазматическим) наследованием, 600000 – аутосомное наследование (информация после 15 мая 1994 г). Агаммаглобулинемия сцепленная с хромосомой X (брутоновская), 300300, ВТК, AGMX 1, IMD 1, XLA, AT, Xq 21. 3 -q 22 Атаксия-телеангиэктазия, 208900, ATM, ATA, ATI, 208900, IIq 22. 3 Болезнь Коудена, 158350, PTEN, MMAC 1, 601728, 10 q 23. 3 Гипериммуноглобулинемия, 144120, Gl, IGHR, 14 q 32. 33 Гломерулонефрит мембранозно пролиферативный, 134370 (фактор Н) HF 1, CFH, HUS, , Iq 32 Дефекты CD: • 186780, CD 3 Z (S-субъединица рецептора Т-лимфоцитов), CD 3 Z, TCRZ, Iq 22 -q 23 • 186940, CD 4+, CD 4, 186940, 12 pter-pl 2 • 107271, CD 59, 11 р13 • 146760, CD 64, FCGR 1 A, IGFR 1, CD 64, Iq 21. 2 -q 21. 3 Дефект адгезии лейкоцитов, 116920, ITGB 2 (интегрин), CD 18, LCAMB, LAD, 600065, 21 q 22. 3 Дефект иммунного интерферона, 147570, IFNG, 12 ql 4 Дефект а-интерферона, 147660, IFNA 1, 9 р22 Дефект лёгких цепей Ig к-Ig, 147200, IGKC, 2 р12 Дефект а-цепи рецептора интерлейкина-2, 147730, IL 2 RA, IL 2 R, 10 р15 -р14 Дефицит инактиватора компонента комплемента 217030, СЗЬ, IF, 4 q 25 Дефицит лактоферрина нейтрофилов, 245480, LTF, 150210 (лактоферрин), 3 q 21 -q 23


; гипоплазия тимуса (синдром Дай") Наиболее часто встречаемые типы врожденного иммунодефицита: тяжелый комбинированный иммунодефицит (ТКИ); гипоплазия тимуса (синдром Дай Джоджа); синдром Незелофа; врожденная агаммаглобулинемия (болезнь Брутона); общий вариабельный (переменный) иммунодефицит; изолированный дефицит Ig. A; иммунодефициты, связанные с наследственными заболеваниями (синдром Вискотта-Олдрича, синдром атаксии-телеангиоэктазии, синдром Блюма) • дефицит комплемента • •
Наиболее часто встречаемые типы врожденного иммунодефицита: тяжелый комбинированный иммунодефицит (ТКИ); гипоплазия тимуса (синдром Дай Джоджа); синдром Незелофа; врожденная агаммаглобулинемия (болезнь Брутона); общий вариабельный (переменный) иммунодефицит; изолированный дефицит Ig. A; иммунодефициты, связанные с наследственными заболеваниями (синдром Вискотта-Олдрича, синдром атаксии-телеангиоэктазии, синдром Блюма) • дефицит комплемента • •
 • • Синдром") Дефицит иммуноглобулинов с высоким или нормальным Ig. M (гипер-Ig. M синдром) • • Синдром представляет группу отдельных заболеваний со сходными клиническими (и фенотипическими) проявлениями. В 70% случаев заболевание наследуется Х-сцепленно, в остальных - аутосомно-рецессивно. Диагностические критерии включают нарушение синтеза антител. Больные могут иметь нормальный Ig. M-антительный ответ, однако, переключение на синтез Ig. G отсутствует. Уровень сывороточного Ig. М (и иногда Ig. D) высокий, в то время как уровени Ig. G и Ig. A снижены. Циркулирующие В лимфоциты несут только Ig. M и Ig. D. Дефект заклю-чается в нарушении переключения синтеза изотипов, однако дефект ДНК в области участка переключения В лимфоцитов отсутствует. У большинства больных отмечается периодическая или постоянная нейтропения и тромбоцитопения. У некоторых выявляются дефекты клеточного звена иммунитета. Генетический дефект, обнаруженный при Х-сцепленной форме, заключается в наличии мутации в гене лиганда CD 40, (CD 40 L) экспрессирующегося на активированных Т лимфоцитах. Взаимодействие CD 40 L и CD 40 на Влимфоцитах необходимо для осуществления переключения синтеза изотипов. Ген лиганда CD 40 локализован на Xq 26, где ранее был картирован генетический дефект при гипер-Ig. M синдроме. CD 40 -лиганд относится ко 2 типу гликопротеидов и гомологичен фактору некроза опухолей. У большинства больных CD 40 -лиганд не экспрессируется на Т-клетках, в некоторых случаях экспрессируется измененный нефункциональный белок, в этом случае, проявления заболевания могут быть менее тяжелыми.
Дефицит иммуноглобулинов с высоким или нормальным Ig. M (гипер-Ig. M синдром) • • Синдром представляет группу отдельных заболеваний со сходными клиническими (и фенотипическими) проявлениями. В 70% случаев заболевание наследуется Х-сцепленно, в остальных - аутосомно-рецессивно. Диагностические критерии включают нарушение синтеза антител. Больные могут иметь нормальный Ig. M-антительный ответ, однако, переключение на синтез Ig. G отсутствует. Уровень сывороточного Ig. М (и иногда Ig. D) высокий, в то время как уровени Ig. G и Ig. A снижены. Циркулирующие В лимфоциты несут только Ig. M и Ig. D. Дефект заклю-чается в нарушении переключения синтеза изотипов, однако дефект ДНК в области участка переключения В лимфоцитов отсутствует. У большинства больных отмечается периодическая или постоянная нейтропения и тромбоцитопения. У некоторых выявляются дефекты клеточного звена иммунитета. Генетический дефект, обнаруженный при Х-сцепленной форме, заключается в наличии мутации в гене лиганда CD 40, (CD 40 L) экспрессирующегося на активированных Т лимфоцитах. Взаимодействие CD 40 L и CD 40 на Влимфоцитах необходимо для осуществления переключения синтеза изотипов. Ген лиганда CD 40 локализован на Xq 26, где ранее был картирован генетический дефект при гипер-Ig. M синдроме. CD 40 -лиганд относится ко 2 типу гликопротеидов и гомологичен фактору некроза опухолей. У большинства больных CD 40 -лиганд не экспрессируется на Т-клетках, в некоторых случаях экспрессируется измененный нефункциональный белок, в этом случае, проявления заболевания могут быть менее тяжелыми.
 Иммунодефициты, связанные с наследственными заболеваниями Синдром Вискотта-Олдрича – наследственное рецессивное заболевание, связанное с Х хромосомой, которое характеризуется экземой, тромбоцитопенией и иммунодефицитом. Дефицит Tлимфоцитов может развиваться в ходе болезни, при этом уровень Ig. M в сыворотке снижен. У больных развиваются рецидивирующие вирусные, грибковые и бактериальные инфекционные болезни, часто возникают лимфомы. Атаксия-телеангиоэктазия – наследственное заболевание, передающееся аутосомно рецессивно, характеризуемое мозжечковой атаксией, телеангиоэктазией кожи и дефицитами Tлимфоцитов, Ig. A и Ig. E. Возможно, что данная патология связанна с наличием дефекта в механизмах репарации ДНК, что приводит к появлению многократных разрывов нитей ДНК, особенно в хромосомах 7 и 11 (гены рецепторов T-клеток). Иногда у данных больных развиваются лимфомы. Синдром Блюма передается аутосомно рецессивно, проявляется в виде других дефектов в репарации ДНК. В клинике наблюдается дефицит иммуноглобулина и часто возникают лимфомы.
Иммунодефициты, связанные с наследственными заболеваниями Синдром Вискотта-Олдрича – наследственное рецессивное заболевание, связанное с Х хромосомой, которое характеризуется экземой, тромбоцитопенией и иммунодефицитом. Дефицит Tлимфоцитов может развиваться в ходе болезни, при этом уровень Ig. M в сыворотке снижен. У больных развиваются рецидивирующие вирусные, грибковые и бактериальные инфекционные болезни, часто возникают лимфомы. Атаксия-телеангиоэктазия – наследственное заболевание, передающееся аутосомно рецессивно, характеризуемое мозжечковой атаксией, телеангиоэктазией кожи и дефицитами Tлимфоцитов, Ig. A и Ig. E. Возможно, что данная патология связанна с наличием дефекта в механизмах репарации ДНК, что приводит к появлению многократных разрывов нитей ДНК, особенно в хромосомах 7 и 11 (гены рецепторов T-клеток). Иногда у данных больных развиваются лимфомы. Синдром Блюма передается аутосомно рецессивно, проявляется в виде других дефектов в репарации ДНК. В клинике наблюдается дефицит иммуноглобулина и часто возникают лимфомы.






 Иммунодефицит: • дефект Т-клеточного рецептора, 186830, CD 3 E, Ilq") Наследственные болезни иммунитета (продолжение) Иммунодефицит: • дефект Т-клеточного рецептора, 186830, CD 3 E, Ilq 23 • недостаточность пурин-нуклеозид фосфорилазы, 164050, NP, 14 ql 3. 1 • Х-сцепленный с повышенным уровнем Ig. M, 308230, CD 40 LG, HIGM 1, IGM, Xq 26 • комбинированный (Х-сцеплениая форма), 312863, 1 L 2 RG (рецептор интерлейкина 2), SCIDX 1, SCIDX, IMD 4, 308380, Xq 13 Иммунодефицит тяжёлый комбинированный: • агаммаглобулинемия швейцарского типа, тип I, 202500 (? ), HYRC 1, DNPK 1, 600899, 8 qll • дефекты интерлейкина 2, 147680, IL 2, 4 q 26 -q 27 • недостаточность аденозиндезаминазы, 102700, ADA, 20 ql 3. 11 • Х-сцепленный, 300400 (рецептор интерлейкина 2), 308380, IL 2 RG, SCIDX 1, SCIDX, IMD 4, Xql 3 • В- и Т-клеточно-негативные формы (недостаточность Янус-киназы), 600173, JAK 3, 19 р13. 1 • В-клеточно-негативная форма, 601457, RAG 1 (179615) и RAG 2 (179616), 11 р13 • дефект Т-клеток, 176947, SRK, ZAP 70, STD, 2 q 12 Недостаточность комплемента: • CR 1, 3 b/4 b рецептор, CR 1, 120620, C 3 BR, Iq 32 ; • C 1 Q, тип В, 120570, C 1 QB, 1 р36. 3 -р34. 1 • C 1 Q, тип С, 120575, C 1 QG, 1 р36. 3 -р34. 1 ; • C 1 Q, тип A, 120550, C 1 QA, 1 р36. 3 -р34. 1 • С 2, 217000, 6 р21. 3. СЗ, 120700, 19 р13. 3 -р13. 2 ; • С 4, С 4 А, C 4 S, 120810, 6 р21. 3 • С 4 В, C 4 F, 120820, 6 р21. 3 ; • С 5, 120900, 9 q 34. 1 • С 6, 217050, 5 р13 ; • С 7, 217070, 5 р13. С 8 А, 120950, 1 р32 • С 9, 120940, 5 р13
Наследственные болезни иммунитета (продолжение) Иммунодефицит: • дефект Т-клеточного рецептора, 186830, CD 3 E, Ilq 23 • недостаточность пурин-нуклеозид фосфорилазы, 164050, NP, 14 ql 3. 1 • Х-сцепленный с повышенным уровнем Ig. M, 308230, CD 40 LG, HIGM 1, IGM, Xq 26 • комбинированный (Х-сцеплениая форма), 312863, 1 L 2 RG (рецептор интерлейкина 2), SCIDX 1, SCIDX, IMD 4, 308380, Xq 13 Иммунодефицит тяжёлый комбинированный: • агаммаглобулинемия швейцарского типа, тип I, 202500 (? ), HYRC 1, DNPK 1, 600899, 8 qll • дефекты интерлейкина 2, 147680, IL 2, 4 q 26 -q 27 • недостаточность аденозиндезаминазы, 102700, ADA, 20 ql 3. 11 • Х-сцепленный, 300400 (рецептор интерлейкина 2), 308380, IL 2 RG, SCIDX 1, SCIDX, IMD 4, Xql 3 • В- и Т-клеточно-негативные формы (недостаточность Янус-киназы), 600173, JAK 3, 19 р13. 1 • В-клеточно-негативная форма, 601457, RAG 1 (179615) и RAG 2 (179616), 11 р13 • дефект Т-клеток, 176947, SRK, ZAP 70, STD, 2 q 12 Недостаточность комплемента: • CR 1, 3 b/4 b рецептор, CR 1, 120620, C 3 BR, Iq 32 ; • C 1 Q, тип В, 120570, C 1 QB, 1 р36. 3 -р34. 1 • C 1 Q, тип С, 120575, C 1 QG, 1 р36. 3 -р34. 1 ; • C 1 Q, тип A, 120550, C 1 QA, 1 р36. 3 -р34. 1 • С 2, 217000, 6 р21. 3. СЗ, 120700, 19 р13. 3 -р13. 2 ; • С 4, С 4 А, C 4 S, 120810, 6 р21. 3 • С 4 В, C 4 F, 120820, 6 р21. 3 ; • С 5, 120900, 9 q 34. 1 • С 6, 217050, 5 р13 ; • С 7, 217070, 5 р13. С 8 А, 120950, 1 р32 • С 9, 120940, 5 р13


 Комбинированные иммунодефициты Наименование Сыв. В Т Предположительный Насле- Ассоци-ироиммуно- клетки патогенез доваванные глобулин ние признаки ы 1 а. Тяжелая N Мутации в ( ) рецепторов Х* комбинированная или IL 2, 4, 7, 9, 15 сцеп иммунная недостаточность Хсцепленная б) Аутосомно. Дефект созревания Т и В АР** рецессивная или N клеток тяжелая комбинированная иммунная недостаточность 2. Дефицит Аномалии Т- и В-клеточные дефекты, АР** аденозиндезаминаз вызванные токсическими хрящей ы (ADA) метаболитами (d. АТФ и Sаденозил гомоцистеином) из -за дефицита фермента 3. Дефицит N N Т-клеточный дефект, АР** Аутоиммунная пуриннуклеозид или вызванный токсическими гемолитическая фосфорилазы (PNP) метаболитами (d. GDP) из-за анемия, дефицита фермента неврологические
Комбинированные иммунодефициты Наименование Сыв. В Т Предположительный Насле- Ассоци-ироиммуно- клетки патогенез доваванные глобулин ние признаки ы 1 а. Тяжелая N Мутации в ( ) рецепторов Х* комбинированная или IL 2, 4, 7, 9, 15 сцеп иммунная недостаточность Хсцепленная б) Аутосомно. Дефект созревания Т и В АР** рецессивная или N клеток тяжелая комбинированная иммунная недостаточность 2. Дефицит Аномалии Т- и В-клеточные дефекты, АР** аденозиндезаминаз вызванные токсическими хрящей ы (ADA) метаболитами (d. АТФ и Sаденозил гомоцистеином) из -за дефицита фермента 3. Дефицит N N Т-клеточный дефект, АР** Аутоиммунная пуриннуклеозид или вызванный токсическими гемолитическая фосфорилазы (PNP) метаболитами (d. GDP) из-за анемия, дефицита фермента неврологические
 4. Дефицит N молекул II Или класса главного комп-лекса HLA 5.") Комбинированные иммунодефициты (продолжение) 4. Дефицит N молекул II Или класса главного комп-лекса HLA 5. Ретикулярная дисгенезия (матери нские) 6. Дефицит CD 3 или CD 3 7. Дефицит CD 8 N N, Мутация факторов транскрипции (генов CD 4 CIITA или RFX-5) для молекул МНС II класса Дефект созревания Т, В лимфоцитов и миелоидных клеток (дефект стволовой клетки) N N Нарушение транскрипции CD 3 или CD 3 цепей N СD 8, Мутации в гене ZAPN 70 киназы CD 4 АР** Задержка развития, затяжная диарея АР** Гранулоцито -пения, тромбоцитопения АР**
Комбинированные иммунодефициты (продолжение) 4. Дефицит N молекул II Или класса главного комп-лекса HLA 5. Ретикулярная дисгенезия (матери нские) 6. Дефицит CD 3 или CD 3 7. Дефицит CD 8 N N, Мутация факторов транскрипции (генов CD 4 CIITA или RFX-5) для молекул МНС II класса Дефект созревания Т, В лимфоцитов и миелоидных клеток (дефект стволовой клетки) N N Нарушение транскрипции CD 3 или CD 3 цепей N СD 8, Мутации в гене ZAPN 70 киназы CD 4 АР** Задержка развития, затяжная диарея АР** Гранулоцито -пения, тромбоцитопения АР**
 Недостаточность лейкотриен С 4 синтетазы, 246530, LTC 4 S, 5") Наследственные болезни иммунитета (продолжение) Недостаточность лейкотриен С 4 синтетазы, 246530, LTC 4 S, 5 q 35 Синдром Вискотта-Олдрича, WAS, 301000, IMD 2, ТНС, Xpll. 23 -pll. 22 Синдром гипериммуноглобулинемии 01, 144120, IGHR, 14 q 32. 33 Синдром голых лимфоцитов • группа комплементации А, 600005, МНС 2 ТА, хр. 16 • группа комплементации С, 601863, RFX 5, Iq 21. 1 -q 21. 3 • тип 1, 170261, ТАР 2, RING 11, PSF 2, 170261, 6 р21. 3 Синдром Ди. Джорджи, 188400, DGCR, DOS, VCF, 22 qll Сочетание синдромов Ди. Джорджи и Шпринтцена, 601362, DGCR 2, DGS 2, 10 р14 -р13 Тромбоцитемия: • эссенциальная, 187950, ТНРО, MGDF, MPLLG, ТРО (тромбопоэтин), 600044, 3 q 26. 3 -q 27 • Х-сцепленная (диатез геморрагический), 313900, WAS, IMD 2, ТНС, • аллоиммунная новорождённых (Гланцманна-Негели болезнь), 273800, ITGA 2 B, GP 2 B, CD 41 B, 17 q 21. 32 • Париж-Трусе 6 (диатезгеморрагический), 188025, ТСРТ, Ilq 23
Наследственные болезни иммунитета (продолжение) Недостаточность лейкотриен С 4 синтетазы, 246530, LTC 4 S, 5 q 35 Синдром Вискотта-Олдрича, WAS, 301000, IMD 2, ТНС, Xpll. 23 -pll. 22 Синдром гипериммуноглобулинемии 01, 144120, IGHR, 14 q 32. 33 Синдром голых лимфоцитов • группа комплементации А, 600005, МНС 2 ТА, хр. 16 • группа комплементации С, 601863, RFX 5, Iq 21. 1 -q 21. 3 • тип 1, 170261, ТАР 2, RING 11, PSF 2, 170261, 6 р21. 3 Синдром Ди. Джорджи, 188400, DGCR, DOS, VCF, 22 qll Сочетание синдромов Ди. Джорджи и Шпринтцена, 601362, DGCR 2, DGS 2, 10 р14 -р13 Тромбоцитемия: • эссенциальная, 187950, ТНРО, MGDF, MPLLG, ТРО (тромбопоэтин), 600044, 3 q 26. 3 -q 27 • Х-сцепленная (диатез геморрагический), 313900, WAS, IMD 2, ТНС, • аллоиммунная новорождённых (Гланцманна-Негели болезнь), 273800, ITGA 2 B, GP 2 B, CD 41 B, 17 q 21. 32 • Париж-Трусе 6 (диатезгеморрагический), 188025, ТСРТ, Ilq 23
 D 82. 1 Синдром Ди Георга, OMIM 188400 Определение") Синдром Ди Джорджи (Ди Георга) D 82. 1 Синдром Ди Георга, OMIM 188400 Определение Синдром Ди Джорджи (СДД) - изолированный Т-клеточный иммунодефицит. Характеризуется триадой ведущих клинических проявлений: гипоплазия тимуса и/или паращитовидных желез и врожденным пороком сердца. В основе СДД лежит порок развития третьего-четвертого глоточных карманов, возникающий между шестой и десятой неделями гестации, приводящий к агенезии или дисгенезии паращитовидных желез и тимуса. Вовлечение первого и второго жаберных карманов приводит к пороку развития лицевых структур, а заинтересованность пятого кармана проявляется широким спектром врожденных пороков сердца с частым вовлечением дуги аорты. Клиническая характеристика: у большинства больных отмечаются диспластические черты лица. Наиболее характерны диспластичные ушные раковины, гипертелоризм, широкая переносица, "рыбий рот", антимонголоидный разрез глаз. У части детей наблюдаются и более грубые аномалии, такие как микрогнатия и незаращение твердого и мягкого неба. Гипокальциемия различной степени тяжести и отсутствие тени вилочковой железы при рентгенографии грудной клетки относятся к частым проявлениям. Гипокальциемические судороги обычно возникают с первых дней жизни. У всех больных отмечается задержка умственного развития. Врожденные пороки сердца и магистральных сосудов также относятся к наиболее характерным и тяжелым признакам заболевания. Иммунологический спектр: количественные показатели Т-клеток варьируют от нормы до глубокой депрессии. Характерна диссоциация между сниженными уровнями Т- и NK-клеток и повышенным
Синдром Ди Джорджи (Ди Георга) D 82. 1 Синдром Ди Георга, OMIM 188400 Определение Синдром Ди Джорджи (СДД) - изолированный Т-клеточный иммунодефицит. Характеризуется триадой ведущих клинических проявлений: гипоплазия тимуса и/или паращитовидных желез и врожденным пороком сердца. В основе СДД лежит порок развития третьего-четвертого глоточных карманов, возникающий между шестой и десятой неделями гестации, приводящий к агенезии или дисгенезии паращитовидных желез и тимуса. Вовлечение первого и второго жаберных карманов приводит к пороку развития лицевых структур, а заинтересованность пятого кармана проявляется широким спектром врожденных пороков сердца с частым вовлечением дуги аорты. Клиническая характеристика: у большинства больных отмечаются диспластические черты лица. Наиболее характерны диспластичные ушные раковины, гипертелоризм, широкая переносица, "рыбий рот", антимонголоидный разрез глаз. У части детей наблюдаются и более грубые аномалии, такие как микрогнатия и незаращение твердого и мягкого неба. Гипокальциемия различной степени тяжести и отсутствие тени вилочковой железы при рентгенографии грудной клетки относятся к частым проявлениям. Гипокальциемические судороги обычно возникают с первых дней жизни. У всех больных отмечается задержка умственного развития. Врожденные пороки сердца и магистральных сосудов также относятся к наиболее характерным и тяжелым признакам заболевания. Иммунологический спектр: количественные показатели Т-клеток варьируют от нормы до глубокой депрессии. Характерна диссоциация между сниженными уровнями Т- и NK-клеток и повышенным
 -") Синдром Вискотта-Олдрича D 82. 0 Синдром Вискотта-Олдрича, OMIM 301000 Определение Синдром Вискотта-Олдрича (СВО) - первичное иммунодефицитное состояние Х-сцепленного типа, проявляющееся триадой симптомов, определяющихся у мальчиков с раннего возраста: • повышенной восприимчивостью к инфекционным заболеваниям • (частые ОРЗ, бронхолегочные инфекции, инфекции ЛОР-органов, • кожи, слизистых, мочевыводящих путей и ЖКТ); • геморрагическим синдромом, обусловленным тромбоцитопенией; • атопическим дерматитом. Для СВО характерны следующие изменения лабораторных показателей: снижение уровня гемоглобина, эритроцитов, тромбоцитов, повышение уровня эозинофилов; изменения уровней сывороточных иммуноглобулинов (низкий Ig. M, нормальный Ig. G, высокий Ig. A, очень высокий Ig. E). Т-клеточные показатели при СВО вариабельны и их интерпрeтация может быть затруднена.
Синдром Вискотта-Олдрича D 82. 0 Синдром Вискотта-Олдрича, OMIM 301000 Определение Синдром Вискотта-Олдрича (СВО) - первичное иммунодефицитное состояние Х-сцепленного типа, проявляющееся триадой симптомов, определяющихся у мальчиков с раннего возраста: • повышенной восприимчивостью к инфекционным заболеваниям • (частые ОРЗ, бронхолегочные инфекции, инфекции ЛОР-органов, • кожи, слизистых, мочевыводящих путей и ЖКТ); • геморрагическим синдромом, обусловленным тромбоцитопенией; • атопическим дерматитом. Для СВО характерны следующие изменения лабораторных показателей: снижение уровня гемоглобина, эритроцитов, тромбоцитов, повышение уровня эозинофилов; изменения уровней сывороточных иммуноглобулинов (низкий Ig. M, нормальный Ig. G, высокий Ig. A, очень высокий Ig. E). Т-клеточные показатели при СВО вариабельны и их интерпрeтация может быть затруднена.

