79602b555ef26c5dbd1c4bd648606f01.ppt
- Количество слайдов: 35



Introduction à la Bioinformatique Alexis Dereeper Bruno Granouillac Burkina Faso, Bobo-dioulasso - 2012

1. Qu’est-ce que la Bioinformatique? Domaine multi-disciplinaire qui utilise des méthodes informatiques (mathématiques, statistiques, combinatoires…) pour résoudre un problème biologique : Formaliser des problèmes de biologie moléculaire; Développer des outils formels; Analyser les données; Prédire des résultats biologiques; Organiser les données. Discipline relativement nouvelle, qui évolue en fonction des nouveaux problèmes posés par la biologie moléculaire. Pas de consensus sur la définition de la bio-informatique.

La bioinformatique s’applique à tout type de données biologiques, en particulier moléculaires : Génome Transcriptome Les séquences d’ADN et de protéines Les structures d’ARN et de protéines Les contenus en gènes des génomes Les puces à ADN (microarrays) Les réseaux d’interactions entre protéines Les réseaux métaboliques Les arbres de phylogénie Utilités : Faire avancer les connaissances en biologie, en génétique humaine, en théorie de l’évolution… Aider à la conception de médicaments Comprendre les maladies complexes. . Protéome Interactome

Quelles sont les données ? Génome Transcriptome Structure 3 D Protéome Interactome Données « haut débit » Evolution

2. Séquençage de génomes Analyser, comprendre et organiser une masse de données biologiques: Plus de 4000 génomes complètement séquencés et publiés, dont l’homme (23 paires de chrom. ) et la souris (20 paires de chrom. ) Projet Hap. Map du génome humain: Construction de la carte des haplotypes Projets de séquençage de milliers de procaryotes et eucaryotes http: //www. genomesonline. org/

Séquençage de génomes Préhistoire De La génomique 454 Life. Science E. coli H. sapiens Solexa Illumina E. coli H. sapiens 2006 2007 2009 2011 20 Mb/run 100 pb 200 000 R. 100 Mb/run 250 pb 400 000 R. 500 Mb/run 500 pb 1 000 R. 800 Mb/run 800 pb 1 500 000 R. 10 X 0, 01 X 1. 5 Gb/run 36 pb 40 000 R. 320 X 0, 5 X 22 X 0, 03 X 3 Gb/run 36 pb 80 000 R. 640 X 1 X 111 X 0, 16 X 10 Gb/run 36 pb 250 000 R. 2000 X 3 X Mais aussi: • Illumina Hi. Seq 200 -> 3 Milliards de lectures (100 pb) • Pac. Bio (Pacific Biosciences) -> X lectures (1000 pb) 266 X 0, 4 X 25 Gb/run 100 pb 250 000 R. >5000 X 9 X

Défis de la bioinformatique Décoder l’information contenue dans les séquences d’ADN et de protéines Trouver les gènes Différencier les introns et les exons (annotation structurale) Analyser les répétitions (SSR, TE…) dans l’ADN Identifier les sites des facteurs de transcription Étudier l’évolution des génomes Génomique structurale: Modéliser les structures 3 D des protéines et des ARN structurels Déterminer la relation entre structure et fonction Génomique fonctionnelle Étudier la régulation des gènes Déterminer les réseaux d’interaction entre les protéines

Assemblage de génome

3. Qu’est-ce qu’un génome? • Des gènes : – portions d’ADN codant des protéines – portions d ’ADN codant des ARN : ARNr, ARNt, ARNsn, … – portions d ’ADN codant des ARN non traduits • Eléments régulateurs : promoteurs, enhancers, … • Eléments requis pour la réplication des chromosomes : origines de réplication, télomères, centromères, … • Séquences non fonctionnelles : – séquences non codantes – séquences répétées – pseudogènes

Taille des génomes Homme Colza Souris Blé Arabidopsis Mais Levure Bacterie Drosophile Riz

Gènes et éléments fonctionnels dans le génome E. coli ? H. sapiens Taille du génome : x 1000 Nombre de gènes : x 10 Gènes protéiques ARN Non codant

Structure de gènes eucaryotes promoteur exons introns site de polyadénylation ADN TRANSCRIPTION préARNm MATURATION signaux d’épissage donneur accepteur AAAAA ATG AG GT AG point de branchement ARNm STOP GT Traduction Régions non traduites (UTR) Régions traduites (CDS) Protéine

Genome Browser

4. Les banques de données bioinformatiques les plus utilisées NCBI, National Center for Biotechnology Information EMBL, The European Molecular Biology Laboratory Ex. PASy, Expert Protein Analysis System, Protéomique Swiss-Prot: Séquences de protéines PROSITE: Domaines et familles de protéines SWISS-MODEL: Outil de prédiction 3 D de protéines Différents outils de recherche PDB, Protein Data Bank Gen. Bank: Séquences d’ADN (3 billion de paires de bases) Site officiel de BLAST Pub. Med: Permet la recherche de références COGs: Familles de gènes orthologues … Base de données de structures 3 D de protéines Visualisation et manipulation de structures SCOP, Structural Classification of Proteins

Les banques de données de séquences biologiques • Une collection de données : – structurées ; – indexées (table des matières) ; – périodiquement mise à jour ; – contenant des références croisées avec d’autres banques. • Il existe essentiellement deux catégories de banques de données : – généralistes : Gen. Bank, EMBL, DDBJ, Swiss. Prot, PIR, … – spécialisées : PDB, Pro. Site, BLOCKS, Pfam, Swiss-3 Dimage, . . .

Structure d’une entrée de la banque de données Identification de la séquence Numéro unique d’accession (Accession Number - AC) Données taxonomiques Références bibliographiques Annotations Références croisées avec d’autres banques de données Mots-clefs ID AC DT DT DT DE DE GN OS OC OC OX RN RP RX RA RA RA RT RT RL CC CC DR DR. . . DR KW KW IL 6_HUMAN STANDARD; PRT; 212 AA. P 05231; 13 -AUG-1987 (Rel. 05, Created) 13 -AUG-1987 (Rel. 05, Last sequence update) 01 -MAR-2002 (Rel. 41, Last annotation update) Interleukin-6 precursor (IL-6) (B-cell stimulatory factor 2) (BSF-2) (Interferon beta-2) (Hybridoma growth factor). IL 6 OR IFNB 2. Homo sapiens (Human). Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Primates; Catarrhini; Hominidae; Homo. NCBI_Tax. ID=9606; [1] SEQUENCE FROM N. A. , AND PARTIAL SEQUENCE. MEDLINE=87065033; Pub. Med=3491322; [NCBI, Ex. PASy, EBI, Israel, Japan] Hirano T. , Yasukawa K. , Harada H. , Taga T. , Watanabe Y. , Matsuda T. , Kashiwamura S. -I. , Nakajima K. , Koyama K. , Iwamatsu A. , Tsunasawa S. , Sakiyama F. , Matsui H. , Takahara Y. , Taniguchi T. , Kishimoto T. ; "Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. "; Nature 324: 73 -76(1986). -!- FUNCTION: IL 6 IS A CYTOKINE WITH A WIDE VARIETY OF BIOLOGICAL FUNCTIONS: IT PLAYS AN ESSENTIAL ROLE IN THE FINAL DIFFERENTIATION OF B-CELLS INTO IG-SECRETING CELLS, IT INDUCES MYELOMA AND PLASMACYTOMA GROWTH, IT INDUCES NERVE CELLS DIFFERENTIATION, IN HEPATOCYTES IT INDUCES ACUTE PHASE REACTANTS. -!- SUBCELLULAR LOCATION: Secreted. -!- SIMILARITY: BELONGS TO THE IL-6 SUPERFAMILY. EMBL; X 04430; CAA 28026. 1; -. [EMBL / Gen. Bank / DDBJ] [Co. Ding. Sequence] EMBL; M 14584; AAA 52728. 1; -. [EMBL / Gen. Bank / DDBJ] [Co. Ding. Sequence] PDB; 1 IL 6; 04 -FEB-98. [Ex. PASy / RCSB] PDB; 2 IL 6; 04 -FEB-98. [Ex. PASy / RCSB] Pfam; PF 00489; IL 6; 1. Cytokine; Glycoprotein; Growth factor; Signal; Polymorphism; 3 D-structure.

Structure d’une entrée de la banque de données Annotation de la séquence FT FT FT FT SQ Séquence Fin de l’entrée SIGNAL CHAIN DISULFID CARBOHYD VARIANT MUTAGEN MUTAGEN SEQUENCE MNSFSTSAFG LDGISALRKE EFEVYLEYLQ AQNQWLQDMT 1 30 72 101 73 32 29 212 78 111 73 32 INTERLEUKIN-6. N-LINKED (GLCNAC. . . ). P -> S. /FTId=VAR_013075. 162 D -> V. /FTId=VAR_013076. 173 A->V: ALMOST NO LOSS OF ACTIVITY. 185 W->R: NO LOSS OF ACTIVITY. 204 S->P: 13% ACTIVITY. 210 R->K, E, Q, T, A, P: LOSS OF ACTIVITY. 212 M->T, N, S, R: LOSS OF ACTIVITY. 212 AA; 23718 MW; 1 F 1 ED 1 FE 1 B 734079 CRC 64; PVAFSLGLLL VLPAAFPAPV PPGEDSKDVA APHRQPLTSS ERIDKQIRYI TCNKSNMCES SKEALAENNL NLPKMAEKDG CFQSGFNEET CLVKIITGLL NRFESSEEQA RAVQMSTKVL IQFLQKKAKN LDAITTPDPT TNASLLTKLQ THLILRSFKE FLQSSLRALR QM // • La séquence peut être formatée : le format FASTA Entrée de Swiss. Prot Numéro unique d’accession Informations diverses (nom, espèce, …) >sp|P 05231|IL 6_HUMAN Interleukin-6 precursor (IL-6) - Homo sapiens (Human). MNSFSTSAFGPVAFSLGLLLVLPAAFPAPVPPGEDSKDVAAPHRQPLTSSERIDKQIRYI LDGISALRKETCNKSNMCESSKEALAENNLNLPKMAEKDGCFQSGFNEETCLVKIITGLL EFEVYLEYLQNRFESSEEQARAVQMSTKVLIQFLQKKAKNLDAITTPDPTTNASLLTKLQ AQNQWLQDMTTHLILRSFKEFLQSSLRALRQM

Les banques de données: accessibilité sur internet • Banques généralistes : • Gen. Bank (Etats-Unis - 1982) : http: //www. ncbi. nlm. nih. gov/Gen. Bank/ • DNA Data. Bank of Japan (Japon - 1986) : http: //www. ddbj. nig. ac. jp • EMBL (Europe - 1980) : http: //www. ebi. ac. uk/embl/ • Banques spécialisées : • Pro. Site : http: //www. expasy. ch/prosite/ • Pfam : http: //www. sanger. ac. uk/Software/Pfam/index. shtml • Brook. Haven Protein Data. Bank (PDB) : http: //www. rcsb. org/pdb/ • Fly. Base : http: //flybase. harvard. edu: 7081/

Recherche dans les bases de données Tache courante d’un biologiste moléculaire Est-ce qu’une nouvelle séquence a déjà été complètement ou partiellement déposée dans les bases de données? Est-ce que cette séquence contient un gène? Est-ce que ce gène appartient à une famille connue? Quelle est la protéine encodée? Existe-t-il d’autres gènes homologues? Existe-t-il des séquences non-codantes similaires. Répétitions ou séquences régulatrices Logiciels les plus connus: Smith-Waterman, FASTA et BLAST

5. Alignement local et global Alignement de deux séquences: Méthodes naturelle pour comparer deux séquences. On compte le nombre de « différences » (insertion, suppression, substitution) • Alignement global (Needlman & Wunsch, 1970) Protéine A Protéine B • Alignement local (Smith & Waterman, 1981 ; FASTA, 1988 ; BLAST, 1990) domaine Protéine A Protéine B ARNm gène

Alignement local: recherche de similarité dans les banques de séquences Pourquoi ? Savoir si ma séquence ressemble à d'autres déjà connues Trouver toutes les séquences d'une même famille Recher toutes les séquences qui contiennent un motif donné Outils grand volume de texte à traiter programmes classiques d’alignement inutilisables utilisation d’heuristiques programmes BLAST et FASTA le résultat n ’est pas garanti comme étant le meilleur

BLAST: Basic Local Alignment Tool • Recherche de régions sans insertions / délétions riches en similarité ; • Détermination d’une longueur de mot : w = 2 ou 3 acides aminés pour les protéines ; • Hachage de la séquence « requête » en mot de taille w Séquence requête m … Liste de mots voisins de longueur w ayant un score supérieur à un seuil T fixé par rapport au mot m. Chaque mot similaire au mot m est comparé à chaque mot de taille w pris dans chaque séquence Bi de la banque. Lorsqu’un mot d’une séquence Bi est identique à un mot de la liste de mots voisins, un hit est enregistré. Pour chaque hit, le programme effectue une extension sans gap de l’alignement dans les deux sens. L’extension s’arrête quand le score du mot étendu diminue de plus qu’un seuil X fixé. Les segments ayant un score de similarité supérieur à un score S seuil fixé sont retenus (High Scoring Pairs = HSP).

BLAST: Choix du programme SEQUENCE BANQUE BLASTP Protéique TB LA ST N ST A BL Protéique X T T BLASTN Nucléique T TBLASTX Nucléique T

On choisit son BLAST La page d’entrée NCBI BLAST http: //www. ncbi. nlm. nih. gov/BLAST/

On entre la séquence à cher

Choisir la banque de données dans laquelle on veut faire la recherche

On a soumis et on attend les résultats

Nombres de hits Répartition des hits en fonction du score



Comparaison de deux génomes

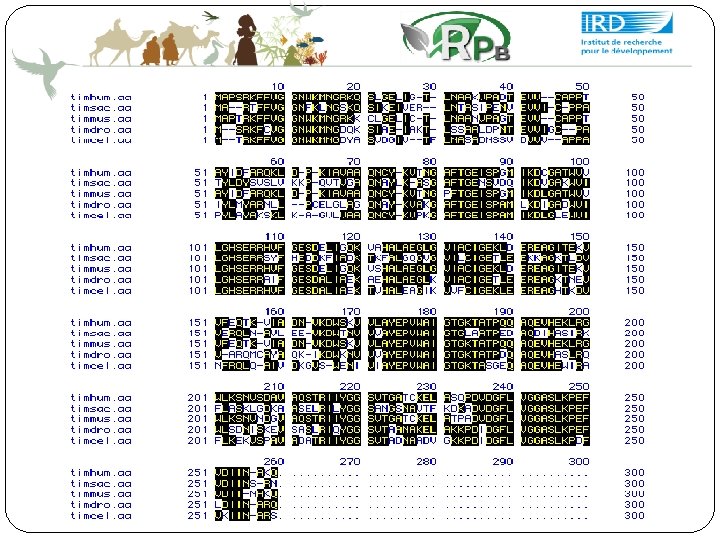
6. Alignement multiple Trouver des caractéristiques communes à une famille de protéines Relier la séquence à la structure et à la fonction Caractériser les gènes homologues Caractériser les régions conservées et les régions variables Déduire des contraintes de structures pour les ARN Construire des arbres de phylogénie

Conservation de régions

Arbres de phylogénie Racine: Ancêtre commun Feuilles: Espèces actuelles Nœuds internes: Points de spéciation Taille des branches: Temps d’évolution
79602b555ef26c5dbd1c4bd648606f01.ppt