

Иммунодефициты.ppt
- Количество слайдов: 55
 Иммунодефициты
Иммунодефициты
 Классификация иммунодефицитов Первичные или врожденные иммунодефициты Вторичные или приобретенные иммунодефициты
Классификация иммунодефицитов Первичные или врожденные иммунодефициты Вторичные или приобретенные иммунодефициты
 D 80 – Иммунодефицит с преобладанием дефектов антител Наследственная гаммаглобулинемия:") Классификация первичных иммунодефицитов (МКБ-10) D 80 – Иммунодефицит с преобладанием дефектов антител Наследственная гаммаглобулинемия: Аутосомно-рецессивный тип (швейцарский тип) Сцепленная с Х-хромосомой агаммаглобулинемия (болезнь Брутона) D 80. 1– несемейная гаммаглобулинемия D 80. 2 – селективный иммунодефицит Ig. A
Классификация первичных иммунодефицитов (МКБ-10) D 80 – Иммунодефицит с преобладанием дефектов антител Наследственная гаммаглобулинемия: Аутосомно-рецессивный тип (швейцарский тип) Сцепленная с Х-хромосомой агаммаглобулинемия (болезнь Брутона) D 80. 1– несемейная гаммаглобулинемия D 80. 2 – селективный иммунодефицит Ig. A
 D 80. 1– несемейная гаммаглобулинемия D 80. 2 – селективный") Классификация первичных иммунодефицитов (МКБ-10) D 80. 1– несемейная гаммаглобулинемия D 80. 2 – селективный иммунодефицит Ig. A D 80. 3 – селективный иммунодефицит Ig. G D 80. 4 – селективный иммунодефицит Ig. M D 80. 5 –иммунодефицит с повышенным уровнем Ig. M D 80. 7 – транзиторная гипогаммаглобулинемия детского возраста
Классификация первичных иммунодефицитов (МКБ-10) D 80. 1– несемейная гаммаглобулинемия D 80. 2 – селективный иммунодефицит Ig. A D 80. 3 – селективный иммунодефицит Ig. G D 80. 4 – селективный иммунодефицит Ig. M D 80. 5 –иммунодефицит с повышенным уровнем Ig. M D 80. 7 – транзиторная гипогаммаглобулинемия детского возраста
 D 81 – комбинированные иммунодефицитные состояния D 81. 1 -") Классификация первичных иммунодефицитов (МКБ-10) D 81 – комбинированные иммунодефицитные состояния D 81. 1 - тяжелый ИД с пониженным количеством Т- и Вклеток D 81. 2 - тяжелый ИД с пониженным и нормальным количеством В-клеток D 81. 3 – недостаточность аденозиндезаминазы D 81. 4 – синдром Незелофа (алимфоцитоз) D 81. 5 – недостаточность пуриннуклеозиддифосфорилазы D 81. 6 – дефицит Аг главного комплекса гистосовместимости класса II и I. Синдром «голых» лимфоцитов. D 81. 8 – дугие виды комбинированных ИД
Классификация первичных иммунодефицитов (МКБ-10) D 81 – комбинированные иммунодефицитные состояния D 81. 1 - тяжелый ИД с пониженным количеством Т- и Вклеток D 81. 2 - тяжелый ИД с пониженным и нормальным количеством В-клеток D 81. 3 – недостаточность аденозиндезаминазы D 81. 4 – синдром Незелофа (алимфоцитоз) D 81. 5 – недостаточность пуриннуклеозиддифосфорилазы D 81. 6 – дефицит Аг главного комплекса гистосовместимости класса II и I. Синдром «голых» лимфоцитов. D 81. 8 – дугие виды комбинированных ИД
 D 82 –ИД в сочетании с другими значительными дефектами D") Классификация первичных иммунодефицитов (МКБ-10) D 82 –ИД в сочетании с другими значительными дефектами D 82. 0 – Синдром Вискотта-Олдрича D 82. 1 – Синдром Ди-Джорджи D 82. 2 – ИД с укорочением конечностей D 82. 4 – Синдром гипергаммаглобулинемии Ig. E D 82. 8 – ИД в сочетании со значительными уточненными дефектами D 84. 1 – Дефекты в системе комплемента
Классификация первичных иммунодефицитов (МКБ-10) D 82 –ИД в сочетании с другими значительными дефектами D 82. 0 – Синдром Вискотта-Олдрича D 82. 1 – Синдром Ди-Джорджи D 82. 2 – ИД с укорочением конечностей D 82. 4 – Синдром гипергаммаглобулинемии Ig. E D 82. 8 – ИД в сочетании со значительными уточненными дефектами D 84. 1 – Дефекты в системе комплемента
 Уровни первичных ИД А. Дефициты специфического звена Дефициты АТ и их компонентов Т-иммунодефициты В. Дефициты неспецифического звена Дефициты системы комплемента Дефициты фагоцитоза
Уровни первичных ИД А. Дефициты специфического звена Дефициты АТ и их компонентов Т-иммунодефициты В. Дефициты неспецифического звена Дефициты системы комплемента Дефициты фагоцитоза
 Классификация вторичных иммунодефицитов По времени возникновения По форме По этиологии По патогенезу
Классификация вторичных иммунодефицитов По времени возникновения По форме По этиологии По патогенезу
 Перинатальные (генотипическая болезнь) Постнатальные (многообразные причины)") Классификация По времени возникновения Антенатальные (ненаследственный синдром Ди-Джорджи) Перинатальные (генотипическая болезнь) Постнатальные (многообразные причины)
Классификация По времени возникновения Антенатальные (ненаследственный синдром Ди-Джорджи) Перинатальные (генотипическая болезнь) Постнатальные (многообразные причины)
 Субкомпенсированная (развитие хронических инфекций) Декомпенсированная (генерализация инфекций") Классификация По форме Компенсированная (часто возникающие инфекции) Субкомпенсированная (развитие хронических инфекций) Декомпенсированная (генерализация инфекций условно-пвтогенной флоры)
Классификация По форме Компенсированная (часто возникающие инфекции) Субкомпенсированная (развитие хронических инфекций) Декомпенсированная (генерализация инфекций условно-пвтогенной флоры)
 Классификация По этиологии Ионизирующая радиация ВИЧ-инфекция Нарушение питания Хирургические операции Болезни обмена Злокачественные опухоли Ожоги Стрессы Возраст Инфекционные заболевания Лекарственные препараты Хроническое течение заболеваний легких, ССС, мочеполовой и др. систем
Классификация По этиологии Ионизирующая радиация ВИЧ-инфекция Нарушение питания Хирургические операции Болезни обмена Злокачественные опухоли Ожоги Стрессы Возраст Инфекционные заболевания Лекарственные препараты Хроническое течение заболеваний легких, ССС, мочеполовой и др. систем
 Классификация По патогенезу Комбинированные Т-клеточный В-клеточный Дефект ЕК –клеток Дефицит макрофагов и гранулоцитов Дефект системы тромбоцитов Дефект системы комплемента
Классификация По патогенезу Комбинированные Т-клеточный В-клеточный Дефект ЕК –клеток Дефицит макрофагов и гранулоцитов Дефект системы тромбоцитов Дефект системы комплемента
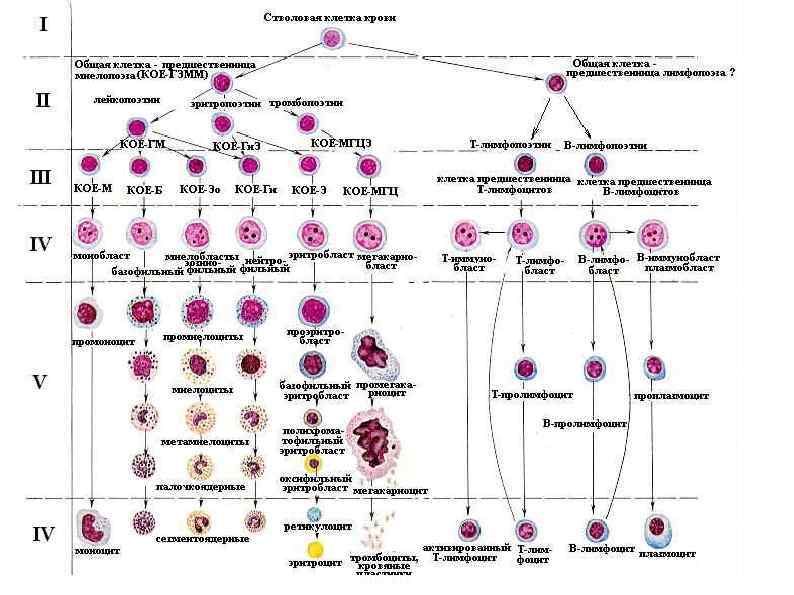
 Комбинированные Синдром лимфоаденопатии – гиперплазии лимфатических узлов Синдром поликлональной активации лимфоцитов") Классификация (по патогенезу) Комбинированные Синдром лимфоаденопатии – гиперплазии лимфатических узлов Синдром поликлональной активации лимфоцитов Сочетание недостаточночти Т- и В- клеток Лимфоцитопения Панлейкопения
Классификация (по патогенезу) Комбинированные Синдром лимфоаденопатии – гиперплазии лимфатических узлов Синдром поликлональной активации лимфоцитов Сочетание недостаточночти Т- и В- клеток Лимфоцитопения Панлейкопения
 Т- клеточный дефект Изолированная Т-лимфоцитопения Дисбаланс Th/Ts (<1, 4) Дефицит цитокинов") Классификация (по патогенезу) Т- клеточный дефект Изолированная Т-лимфоцитопения Дисбаланс Th/Ts (<1, 4) Дефицит цитокинов (Ил-2, ИНФ и др. )
Классификация (по патогенезу) Т- клеточный дефект Изолированная Т-лимфоцитопения Дисбаланс Th/Ts (<1, 4) Дефицит цитокинов (Ил-2, ИНФ и др. )
 В-клеточный дефект В-клеточные опухоли Панцитогаммаглобулинемия Дисгаммаглобулинемия Синдром дефицита антите Дефицит секреторного") Классификация (по патогенезу) В-клеточный дефект В-клеточные опухоли Панцитогаммаглобулинемия Дисгаммаглобулинемия Синдром дефицита антите Дефицит секреторного Ig. A Дефицит субклассов Ig. G – Ig. G 1, Ig. G 2, Ig. G 3, Ig. G 4 Дефицит метаболической активности нейтрофилов – снижение НСТ
Классификация (по патогенезу) В-клеточный дефект В-клеточные опухоли Панцитогаммаглобулинемия Дисгаммаглобулинемия Синдром дефицита антите Дефицит секреторного Ig. A Дефицит субклассов Ig. G – Ig. G 1, Ig. G 2, Ig. G 3, Ig. G 4 Дефицит метаболической активности нейтрофилов – снижение НСТ
 Дефект ЕК –клеток Выявляется при достоверном снижении ЕК Дефект системы тромбоцитов") Классификация (по патогенезу) Дефект ЕК –клеток Выявляется при достоверном снижении ЕК Дефект системы тромбоцитов Тромбоцитопенический синдром Дефект системы комплемента Гипокомплементемия
Классификация (по патогенезу) Дефект ЕК –клеток Выявляется при достоверном снижении ЕК Дефект системы тромбоцитов Тромбоцитопенический синдром Дефект системы комплемента Гипокомплементемия
 Дефицит макрофагов и гранулоцитов Синдром ВИД при миелозах Синдром гиперактивации макрофагов-моноцитов") Классификация (по патогенезу) Дефицит макрофагов и гранулоцитов Синдром ВИД при миелозах Синдром гиперактивации макрофагов-моноцитов Панфанцитопения Синдром гмперэозинофилии Дефицит рецепторов нейтрофилов и молекул адгезии Дефицит хемотаксической активности нейтрофилов Дефицит поглотительной активности нейтрофилов
Классификация (по патогенезу) Дефицит макрофагов и гранулоцитов Синдром ВИД при миелозах Синдром гиперактивации макрофагов-моноцитов Панфанцитопения Синдром гмперэозинофилии Дефицит рецепторов нейтрофилов и молекул адгезии Дефицит хемотаксической активности нейтрофилов Дефицит поглотительной активности нейтрофилов
 ТКИД, синдром Вискотта-Олдрича, синдром Ди Джорджи, хронический слизисто-кожный") Первичные иммунодефициты Недостаточность клеточного иммунитета (Тиммунодефицит) ТКИД, синдром Вискотта-Олдрича, синдром Ди Джорджи, хронический слизисто-кожный кандидоз, синдром Незелофа, Синдром Луи-Барра, ретикулярная дисгенезия, вариабельные ИД
Первичные иммунодефициты Недостаточность клеточного иммунитета (Тиммунодефицит) ТКИД, синдром Вискотта-Олдрича, синдром Ди Джорджи, хронический слизисто-кожный кандидоз, синдром Незелофа, Синдром Луи-Барра, ретикулярная дисгенезия, вариабельные ИД
 агаммаглобулинемия (болезнь Брутона), селективный дефицит Ig. A") Первичные иммунодефициты Недостаточность гуморального иммунного ответа (В-иммунодефицит) агаммаглобулинемия (болезнь Брутона), селективный дефицит Ig. A и его субклассов, дефицит Ig. G и Ig. A, дефицит Ig. M и Ig. A, транзиторная младенческая гипогаммаглобулинемия, кишечная лимфаангиэктазия
Первичные иммунодефициты Недостаточность гуморального иммунного ответа (В-иммунодефицит) агаммаглобулинемия (болезнь Брутона), селективный дефицит Ig. A и его субклассов, дефицит Ig. G и Ig. A, дефицит Ig. M и Ig. A, транзиторная младенческая гипогаммаглобулинемия, кишечная лимфаангиэктазия
, дефицит С") Первичные иммунодефициты Наследственная патология системы комплемента ангионевротический отек (недостаточность ингибитора Cq 1), дефицит С 1, дефицит С 2, дефицит С 3, дефицит С 4; недостаточность поздних компонентов комплемента (С 5, С 6, С 7, С 8), недостаточность белков альтернативного пути активации (фактора Н, фактора I и пропердина)
Первичные иммунодефициты Наследственная патология системы комплемента ангионевротический отек (недостаточность ингибитора Cq 1), дефицит С 1, дефицит С 2, дефицит С 3, дефицит С 4; недостаточность поздних компонентов комплемента (С 5, С 6, С 7, С 8), недостаточность белков альтернативного пути активации (фактора Н, фактора I и пропердина)
, дефицит факторов киллинга, синдром") Первичные иммунодефициты Нарушения системы фагоцитоза дефициты хемотаксиса (синдром «ленивых» лейкоцитов), дефицит факторов киллинга, синдром Чедиака-Хигаси, синдром Джоба (Йова), наследственный хронический агранулоцитоз, периодическая циклическая нейтропения.
Первичные иммунодефициты Нарушения системы фагоцитоза дефициты хемотаксиса (синдром «ленивых» лейкоцитов), дефицит факторов киллинга, синдром Чедиака-Хигаси, синдром Джоба (Йова), наследственный хронический агранулоцитоз, периодическая циклическая нейтропения.
 Первичные иммунодефициты
Первичные иммунодефициты
 Первичные иммунодефициты
Первичные иммунодефициты

 ТКИД * * Х-сцепленный тип • Специфический дефект: • Нарушение дифференцировки стволовой клетки в В - и Т-лимфоциты. • Дефект гамма-цепи рецептора к Ил 2 на Т -лф
ТКИД * * Х-сцепленный тип • Специфический дефект: • Нарушение дифференцировки стволовой клетки в В - и Т-лимфоциты. • Дефект гамма-цепи рецептора к Ил 2 на Т -лф
 ТКИД Аутосомно-рецессивный тип Специфический дефект: Мутация гена тирозинкиназы ZAP-70 (трансдуктор сигнала в Т-лф для их пролиферации) Отсутствие CD 8+ клеток в крови Локализация в хромосоме Xq 13 -21. 1
ТКИД Аутосомно-рецессивный тип Специфический дефект: Мутация гена тирозинкиназы ZAP-70 (трансдуктор сигнала в Т-лф для их пролиферации) Отсутствие CD 8+ клеток в крови Локализация в хромосоме Xq 13 -21. 1
 Т-комбинированный иммунодефицит В раннем возрасте инфекции: затяжная диаррея, пневмония, кандидозы. Иммунизация вакцинами приводит к развитию заболевания Дети погибают в течение 2 лет В крови мало лимфоцитов (<3000/мл)
Т-комбинированный иммунодефицит В раннем возрасте инфекции: затяжная диаррея, пневмония, кандидозы. Иммунизация вакцинами приводит к развитию заболевания Дети погибают в течение 2 лет В крови мало лимфоцитов (<3000/мл)
 Синдром Ди Джорджи Гипо-, аплазия тимуса Специфический дефект: Дисэмбриогенез (нарушение развития тимуса, щитовидной и паращитовидной желез) Снижение количества и функции Т-лф Снижение способности продуцировать Ат Локализация в хромосоме 22 q. II
Синдром Ди Джорджи Гипо-, аплазия тимуса Специфический дефект: Дисэмбриогенез (нарушение развития тимуса, щитовидной и паращитовидной желез) Снижение количества и функции Т-лф Снижение способности продуцировать Ат Локализация в хромосоме 22 q. II
 Синдром Вискотта-Олдрича ** * • Специфический дефект • Нарушена активация CD 4+ и CD 8+ клеток. • Нарушение продукции Ig. M к капсульным бактериям • Повышены Ig. А и Ig. Е • Изогемагглютинины снижены или отсутствуют • Локализация в хромосоме Xp. 11 -11. 3 (WASP)
Синдром Вискотта-Олдрича ** * • Специфический дефект • Нарушена активация CD 4+ и CD 8+ клеток. • Нарушение продукции Ig. M к капсульным бактериям • Повышены Ig. А и Ig. Е • Изогемагглютинины снижены или отсутствуют • Локализация в хромосоме Xp. 11 -11. 3 (WASP)
 Специфический дефект Нарушения функции Т-и В-лимфоцитов Снижение уровня Ig. А, Ig.") Синдром Луи-Барр (атаксиятелеангиэктазия) Специфический дефект Нарушения функции Т-и В-лимфоцитов Снижение уровня Ig. А, Ig. Е и Ig. G 2 Гипоплазия тимуса, селезенки, лимфоузлов и миндалин. Локализация в хромосоме IIq 22. 3(atm)
Синдром Луи-Барр (атаксиятелеангиэктазия) Специфический дефект Нарушения функции Т-и В-лимфоцитов Снижение уровня Ig. А, Ig. Е и Ig. G 2 Гипоплазия тимуса, селезенки, лимфоузлов и миндалин. Локализация в хромосоме IIq 22. 3(atm)
 ИД с повышенным содержанием Ig. M * • Специфический дефект • Отсутствие на Т-х CD 40 лиганда. • Нарушено взаимодействие Т-и В-лф • Отсутствует переключение синтеза иммуноглобулинов с Ig. M на Ig. G и других классов • Отсутствие формирования соотвествующих АОК плазматических клеток. • Низкие уровни Ig. G, А и Е. • Локализация в хромосоме Xq 26. 27
ИД с повышенным содержанием Ig. M * • Специфический дефект • Отсутствие на Т-х CD 40 лиганда. • Нарушено взаимодействие Т-и В-лф • Отсутствует переключение синтеза иммуноглобулинов с Ig. M на Ig. G и других классов • Отсутствие формирования соотвествующих АОК плазматических клеток. • Низкие уровни Ig. G, А и Е. • Локализация в хромосоме Xq 26. 27
 • • * Специфический дефект Отсутствие В-клеток Низкие уровни Ig Дефект") Агаммаглобулинемия (болезнь Брутона) • • * Специфический дефект Отсутствие В-клеток Низкие уровни Ig Дефект гена кодирующего тирозинкиназу (Btk) (Трансдуктор активации и дифференцировки В-л в АОК) • Локализация в хромосоме Xq 21. 322(b+k)
Агаммаглобулинемия (болезнь Брутона) • • * Специфический дефект Отсутствие В-клеток Низкие уровни Ig Дефект гена кодирующего тирозинкиназу (Btk) (Трансдуктор активации и дифференцировки В-л в АОК) • Локализация в хромосоме Xq 21. 322(b+k)
 Общая вариабельная гипогаммаглобулинемия Специфический дефект: Снижение уровня Ig. M, Ig. G, Ig. A. В-лимфоциты в норме или несколько снижены Дефицит антителообразования Дефекты Т-звена Локализация дефекта: в хромосоме 6 р21. 3
Общая вариабельная гипогаммаглобулинемия Специфический дефект: Снижение уровня Ig. M, Ig. G, Ig. A. В-лимфоциты в норме или несколько снижены Дефицит антителообразования Дефекты Т-звена Локализация дефекта: в хромосоме 6 р21. 3
 Специфический дефект Низкие уровни иммунолобулинов") Транзиторная гипогаммаглобулинемия (медленный иммунологический старт) Специфический дефект Низкие уровни иммунолобулинов
Транзиторная гипогаммаглобулинемия (медленный иммунологический старт) Специфический дефект Низкие уровни иммунолобулинов
 Специфический дефект Снижение уровня иммуноглобулинов (нарушение переключение синтеза с Ig.") Дисгаммаглобулинемия (избирательный дефицит Ig) Специфический дефект Снижение уровня иммуноглобулинов (нарушение переключение синтеза с Ig. G на Ig. A) Иммуно-лабораторное исследование: Следы Ig. A при нормальном содержании Ig. G Нормальный или повышенный уровень Ig. М Снижение Т-х и повышение Т-с
Дисгаммаглобулинемия (избирательный дефицит Ig) Специфический дефект Снижение уровня иммуноглобулинов (нарушение переключение синтеза с Ig. G на Ig. A) Иммуно-лабораторное исследование: Следы Ig. A при нормальном содержании Ig. G Нормальный или повышенный уровень Ig. М Снижение Т-х и повышение Т-с
 Иммунодефицит системы комплемента Компоненты С” Клинические проявления C 1 q C 1 r Высокая частота ИК патологии (СКВ, гломерулонефрит) То же C 2 То же C 4 То же C 3 Рецидивирующая пиогенная инфекция C 5 Рецидивирующая гонококковая инфекция, высокая частота СКВ Рецидивирующая гонококковая инфекция C 6
Иммунодефицит системы комплемента Компоненты С” Клинические проявления C 1 q C 1 r Высокая частота ИК патологии (СКВ, гломерулонефрит) То же C 2 То же C 4 То же C 3 Рецидивирующая пиогенная инфекция C 5 Рецидивирующая гонококковая инфекция, высокая частота СКВ Рецидивирующая гонококковая инфекция C 6
 Иммунодефицит системы комплемента Компоненты С” Клинические проявления C 7 Рецидивирующая гонококковая инфекция C 8 То же C 9 Протекает асимптоматически C 1 - Ангионевротический отек ингибитор Фактор 1 (C 3 b инактиватор) Рецидивирующая пиогенная инфекция Фактор Н Рецидивирующая пиогенная инфекция Пропердин Рецидивирующая гонококковая инфекция
Иммунодефицит системы комплемента Компоненты С” Клинические проявления C 7 Рецидивирующая гонококковая инфекция C 8 То же C 9 Протекает асимптоматически C 1 - Ангионевротический отек ингибитор Фактор 1 (C 3 b инактиватор) Рецидивирующая пиогенная инфекция Фактор Н Рецидивирующая пиогенная инфекция Пропердин Рецидивирующая гонококковая инфекция
") Иммунодефицит системы комплемента Недостаточность С 1 ингибитора Выделяют 2 формы: Истинный НАО (функция сохранена) Вариантный НАО (функция резко снижена)
Иммунодефицит системы комплемента Недостаточность С 1 ингибитора Выделяют 2 формы: Истинный НАО (функция сохранена) Вариантный НАО (функция резко снижена)
 Патогенез НАО С 1 -ингибитор блокирует классический путь активации С. Подавляет активность кининовой и плазминовой систем Системы свертывания крови. Происходит накопление пептидов С 5 а и С 3 а (сильное вазотропное действие и повышение проницаемости капилляров) Нерегулируемая активация всех процессов ведет к образованию брадикинина и кинина С 2, вызывающих отеки.
Патогенез НАО С 1 -ингибитор блокирует классический путь активации С. Подавляет активность кининовой и плазминовой систем Системы свертывания крови. Происходит накопление пептидов С 5 а и С 3 а (сильное вазотропное действие и повышение проницаемости капилляров) Нерегулируемая активация всех процессов ведет к образованию брадикинина и кинина С 2, вызывающих отеки.
 Дефицит системы фагоцитов Состояние Клиника Дефект Тип наследования Дефицит молекул адгезии 1. Задержка отделения последа; инфекции кожи; гингивиты; абсцессы, перитониты, остеомиелиты Неспособность адгезии лейкоцитов к эпителию и миграции в очаг воспаления и осуществлять фагоцитоз бактерий Аутосомно рецессивный Синдром Чедиака -Хигаси Увеличение размера лизосом «гигантские гранулы» ; частичный альбинизм Нарушение хемотаксиса нейтрофилов, снижение микробицидной активности, из-за неспособности слияния фаго-и лизосомы Аутосомно рецессивный Гипер-Ig. Eсиндром Кандидоз кожи и слизистых; высокий уровень Ig. Е; абсцессы легких; нарушение кальциевого обмена Нарушения хемотаксиса Аутосомно рецессивный Хроническая Абсцессы, вызываемые Снижение образования Аутосомно гранулематозная микроорганизмами продуктов «дыхательного рецессивный болезнь содержащими каталазу, взрыва» и неспособность к образование гранулем киллингу стафилококков и грибов
Дефицит системы фагоцитов Состояние Клиника Дефект Тип наследования Дефицит молекул адгезии 1. Задержка отделения последа; инфекции кожи; гингивиты; абсцессы, перитониты, остеомиелиты Неспособность адгезии лейкоцитов к эпителию и миграции в очаг воспаления и осуществлять фагоцитоз бактерий Аутосомно рецессивный Синдром Чедиака -Хигаси Увеличение размера лизосом «гигантские гранулы» ; частичный альбинизм Нарушение хемотаксиса нейтрофилов, снижение микробицидной активности, из-за неспособности слияния фаго-и лизосомы Аутосомно рецессивный Гипер-Ig. Eсиндром Кандидоз кожи и слизистых; высокий уровень Ig. Е; абсцессы легких; нарушение кальциевого обмена Нарушения хемотаксиса Аутосомно рецессивный Хроническая Абсцессы, вызываемые Снижение образования Аутосомно гранулематозная микроорганизмами продуктов «дыхательного рецессивный болезнь содержащими каталазу, взрыва» и неспособность к образование гранулем киллингу стафилококков и грибов
 ИД фагоцитоза Хронический гранулематоз Специфический дефект: Нарушение переваривающей активности нейтрофилов из-за нарушения кислородзависимого метаболизма, снижения активности НАДФоксидазы Локализация в хромосоме 1 q 25
ИД фагоцитоза Хронический гранулематоз Специфический дефект: Нарушение переваривающей активности нейтрофилов из-за нарушения кислородзависимого метаболизма, снижения активности НАДФоксидазы Локализация в хромосоме 1 q 25
 Вторичные иммунодефициты
Вторичные иммунодефициты
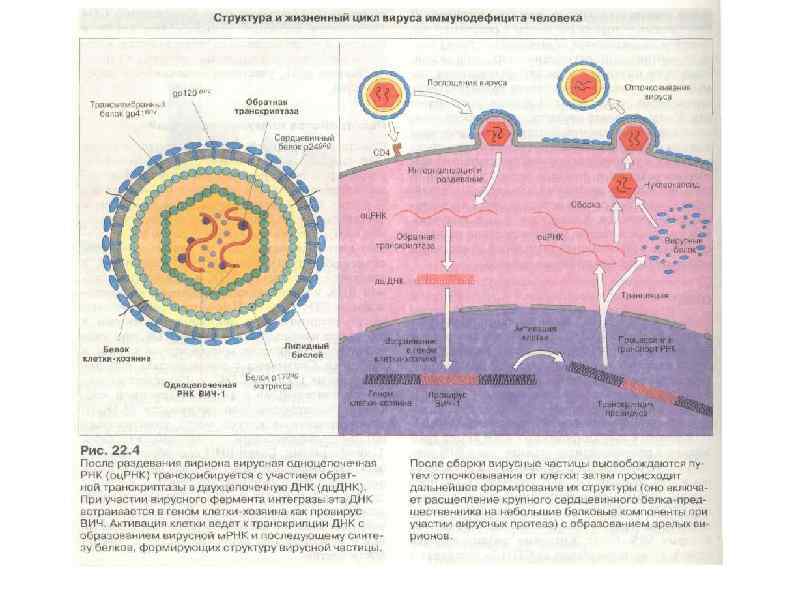
") ВИЧ-инфекция (СПИД)
ВИЧ-инфекция (СПИД)
 СПИД
СПИД
 звена иммунитета Оценка гуморального (В-)") Иммуно-лабораторное обследование ОАК, СОЭ, С-реактивный белок Оценка клеточного (Т-) звена иммунитета Оценка гуморального (В-) звена иммунитета Оценка системы фагоцитоза Оценка системы комплемента При необходимости: Количество и функция ЕК-клеток (CD 16/CD 56) HLA – фенотип Продукция провоспалительных цитокинов (Ил 2, γИФ, α-ФНО, Ил 8, Ил 12) Продукция противовоспалительных цитокинов (Ил 4, Ил 5, Ил 10, Ил 13) Наличие специфических аутоантител Наличие специфической клеточной сенсибелизации Наличие Т-и В-лимфоцитов с признаками активации (DR, CD 25, CD 71)
Иммуно-лабораторное обследование ОАК, СОЭ, С-реактивный белок Оценка клеточного (Т-) звена иммунитета Оценка гуморального (В-) звена иммунитета Оценка системы фагоцитоза Оценка системы комплемента При необходимости: Количество и функция ЕК-клеток (CD 16/CD 56) HLA – фенотип Продукция провоспалительных цитокинов (Ил 2, γИФ, α-ФНО, Ил 8, Ил 12) Продукция противовоспалительных цитокинов (Ил 4, Ил 5, Ил 10, Ил 13) Наличие специфических аутоантител Наличие специфической клеточной сенсибелизации Наличие Т-и В-лимфоцитов с признаками активации (DR, CD 25, CD 71)
 Диагностика ИД
Диагностика ИД
 Иммунодиагностика
Иммунодиагностика
 Иммунодиагностика
Иммунодиагностика
 Иммунодиагностика
Иммунодиагностика
 Иммунодиагностика
Иммунодиагностика
 Иммунодиагностика
Иммунодиагностика
 Иммунодиагностика
Иммунодиагностика
 Трансгенные животные
Трансгенные животные