Анемии гемолитич A.ppt
- Количество слайдов: 107
 Hemolytic anemia – increased destruction of erythrocytes
Hemolytic anemia – increased destruction of erythrocytes
 Hemolytic anemia – increased destruction of erythrocytes with adequate response of bone marrow.
Hemolytic anemia – increased destruction of erythrocytes with adequate response of bone marrow.








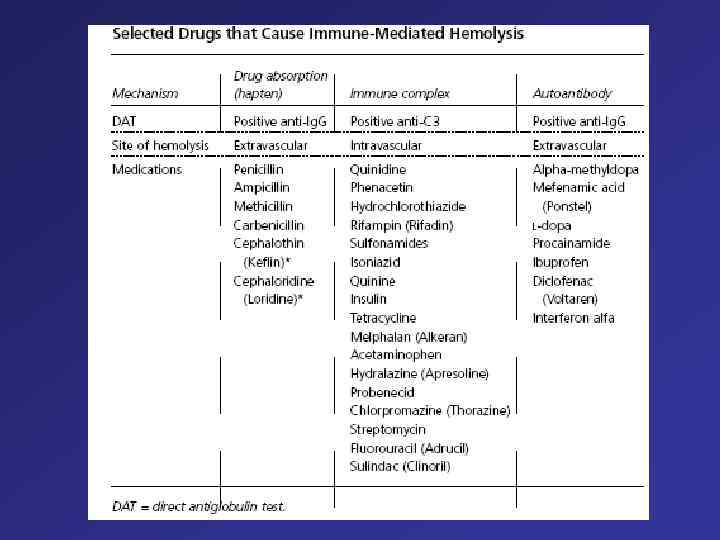
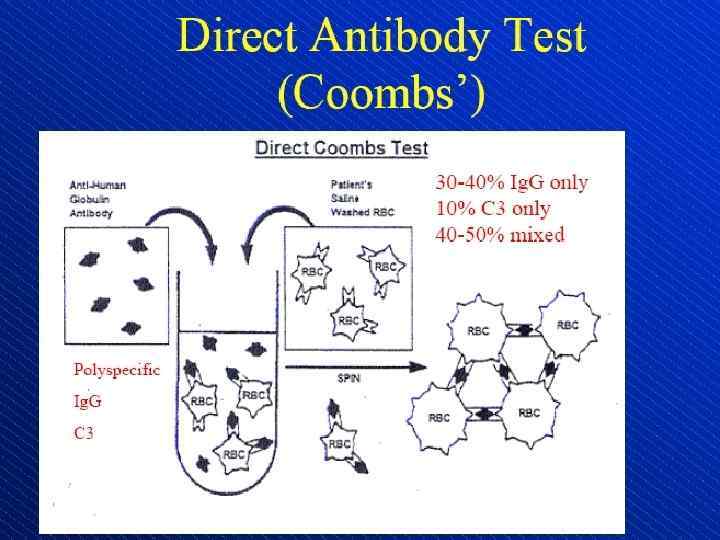
 or complement on the") Direct antiglobulin test, demonstrating the presence of autoantibodies (shown here) or complement on the surface of the red blood cell. (RBCs = red blood cells)
Direct antiglobulin test, demonstrating the presence of autoantibodies (shown here) or complement on the surface of the red blood cell. (RBCs = red blood cells)






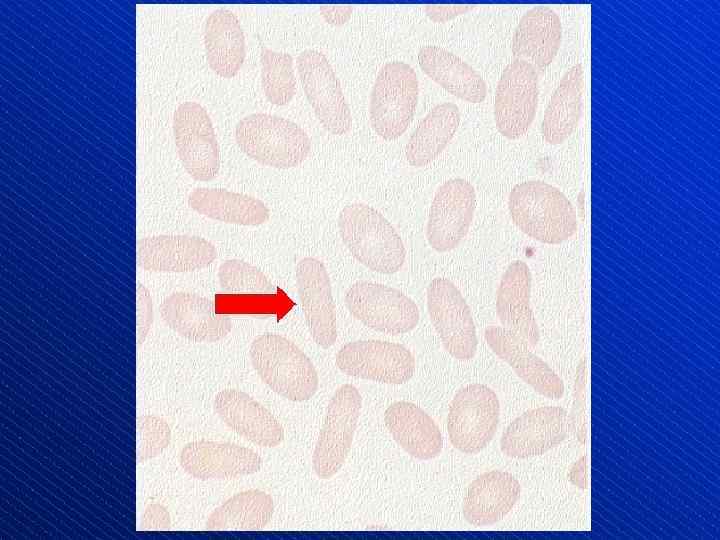
") Schistocytes (arrows)
Schistocytes (arrows)






















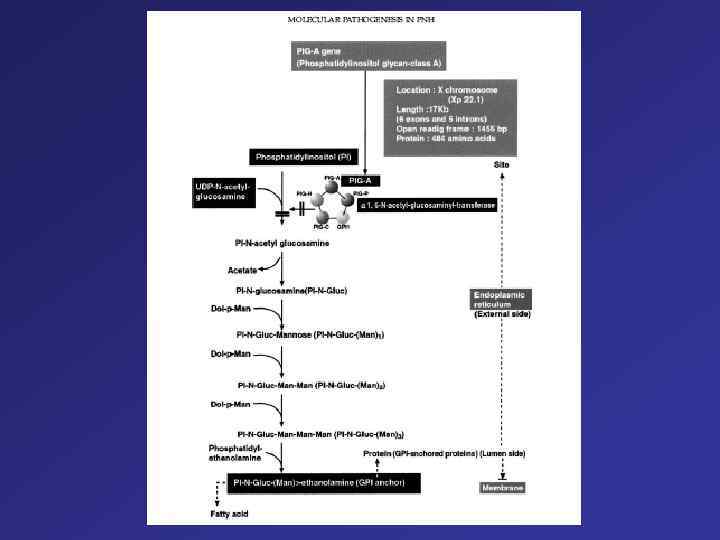
 = anchore for different molecules Mutations – Chromosome X (PIG-Agene) Deletions") PNH Glucosylphosphalidylinosinto (GPI) = anchore for different molecules Mutations – Chromosome X (PIG-Agene) Deletions 70 % Insertion Point mutations Loss Function Loss of GPI anchored proteins (F. i. CD 55, CD 59 – prevention of complemented-mediated lyses of sells) Anemia, Cytopenia
PNH Glucosylphosphalidylinosinto (GPI) = anchore for different molecules Mutations – Chromosome X (PIG-Agene) Deletions 70 % Insertion Point mutations Loss Function Loss of GPI anchored proteins (F. i. CD 55, CD 59 – prevention of complemented-mediated lyses of sells) Anemia, Cytopenia
 PNH Sleeping Hypoventilation Acidofication of Plasma Complement + Activation Increased Hemolysis
PNH Sleeping Hypoventilation Acidofication of Plasma Complement + Activation Increased Hemolysis
") Venous thrombotic events Deficiency of the receptor for urokinase type plasminogen activator (u. PAR) Increased soluble u. PAR
Venous thrombotic events Deficiency of the receptor for urokinase type plasminogen activator (u. PAR) Increased soluble u. PAR
") Target cells (arrows)
Target cells (arrows)
. Step 1, somatic mutation") Three-step model for the development of paroxysmal nocturnal haemoglobinuria (PNH). Step 1, somatic mutation of phosphatidylinositolglycan (PIG-A) in a haemopoietic stem cell. Step 2, immunological attack on haemopoietic stem cells selectively decreases the number of PIG -normal stem cells, resulting in relative increase in the PIG-A mutant clone. Step 3, a second mutation occurs in the PIG-A mutant, leading to further expansion and generation of a large number of PIG-deficient blood cells.
Three-step model for the development of paroxysmal nocturnal haemoglobinuria (PNH). Step 1, somatic mutation of phosphatidylinositolglycan (PIG-A) in a haemopoietic stem cell. Step 2, immunological attack on haemopoietic stem cells selectively decreases the number of PIG -normal stem cells, resulting in relative increase in the PIG-A mutant clone. Step 3, a second mutation occurs in the PIG-A mutant, leading to further expansion and generation of a large number of PIG-deficient blood cells.
 Relationship between bone marrow failure syndromes, including AA, PNH and MDS, and acute leukemia. Young proposed bone marrow failure syndromes that arise from a common feature of hypoplastic or aplastic bone marrow and may underly clonal hematopoiesis
Relationship between bone marrow failure syndromes, including AA, PNH and MDS, and acute leukemia. Young proposed bone marrow failure syndromes that arise from a common feature of hypoplastic or aplastic bone marrow and may underly clonal hematopoiesis
 Occurrence and expansion of a PNH clone and the clinical course of PNH.
Occurrence and expansion of a PNH clone and the clinical course of PNH.
 Population distribution of prevalent b-thalassemia mutations
Population distribution of prevalent b-thalassemia mutations
 Timing of the normal developmental switching of human hemoglobin Early in fetal life the synthesis of the embryonic -globin–like ( ) chains switches to that of -globin, which is produced thereafter. At the same time, the synthesis of embryonic beta-like ( chains switches to that of globin chains. The -globin and -globin chains combine to form fetal hemoglobin ( 2 2), the main ß-globin– like globin during the remainder of fetal life and throughout early postnatal life. As a result of the decline in the synthesis of -globin chains in patients with ß-thalassemia, fetal hemoglobin production becomes insufficient to compensate for the excess of -globin chains, the production of which is unaffected in ß-thalassemia. )
Timing of the normal developmental switching of human hemoglobin Early in fetal life the synthesis of the embryonic -globin–like ( ) chains switches to that of -globin, which is produced thereafter. At the same time, the synthesis of embryonic beta-like ( chains switches to that of globin chains. The -globin and -globin chains combine to form fetal hemoglobin ( 2 2), the main ß-globin– like globin during the remainder of fetal life and throughout early postnatal life. As a result of the decline in the synthesis of -globin chains in patients with ß-thalassemia, fetal hemoglobin production becomes insufficient to compensate for the excess of -globin chains, the production of which is unaffected in ß-thalassemia. )
 α 2 β 2 Adult (A)") Types of Hemoglobin α 2γ 2 Fetal (F) α 2 β 2 Adult (A) α 2δ 2 Adult (A 2) α - chromosome 16 β, γ - chromosome 11
Types of Hemoglobin α 2γ 2 Fetal (F) α 2 β 2 Adult (A) α 2δ 2 Adult (A 2) α - chromosome 16 β, γ - chromosome 11
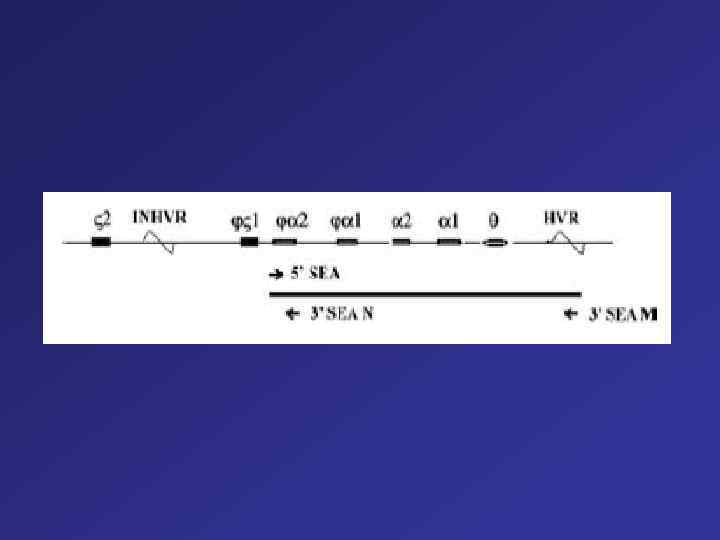
 The ß-Globin Gene Cluster on the Short Arm of Chromosome 11 ß-globin–like genes are arranged in the order in which they are expressed during development. The G and A genes are both active genes that produce -globin chains that differ only at position 136 (glycine is the product of the G gene, and alanine is the product of the A gene). The ß gene is a pseudogene, an evolutionary remnant of a previously active ß-globin–like gene. Areas of nucleotide homology upstream from the initiation codon of each active gene, termed promoter elements, are involved in the initiation of transcription and hence play a vital part in gene regulation. The cluster also contains regulatory elements that interact to promote erythroid-specific gene expression and to coordinate the developmental regulation of each gene, including hemoglobin switching. These include enhancers, distant regulatory elements that increase gene expression, and a master sequence, the cluster's essential distal regulatory element, the ßglobin locus-control region. This is a region that lies 20 kb upstream from the globin gene. 13 It encompasses five erythroid-lineage–specific nuclease hypersensitive sites (shown in red) that permit expression of the downstream genes. In addition to these elements for up-regulation, several suppressor regions, or silencers, have been defined in the ßglobin gene cluster. -
The ß-Globin Gene Cluster on the Short Arm of Chromosome 11 ß-globin–like genes are arranged in the order in which they are expressed during development. The G and A genes are both active genes that produce -globin chains that differ only at position 136 (glycine is the product of the G gene, and alanine is the product of the A gene). The ß gene is a pseudogene, an evolutionary remnant of a previously active ß-globin–like gene. Areas of nucleotide homology upstream from the initiation codon of each active gene, termed promoter elements, are involved in the initiation of transcription and hence play a vital part in gene regulation. The cluster also contains regulatory elements that interact to promote erythroid-specific gene expression and to coordinate the developmental regulation of each gene, including hemoglobin switching. These include enhancers, distant regulatory elements that increase gene expression, and a master sequence, the cluster's essential distal regulatory element, the ßglobin locus-control region. This is a region that lies 20 kb upstream from the globin gene. 13 It encompasses five erythroid-lineage–specific nuclease hypersensitive sites (shown in red) that permit expression of the downstream genes. In addition to these elements for up-regulation, several suppressor regions, or silencers, have been defined in the ßglobin gene cluster. -
 Pathophysiology of Sickle Cell Disease In hemoglobin S, a substitution of T for A in the sixth codon of the ßglobin gene leads to the replacement of a glutamic acid residue by a valine residue. On deoxygenation, hemoglobin S polymers form, causing cell sickling and damage to the membrane. Some sickle cells adhere to endothelial cells, leading to vasoocclusion.
Pathophysiology of Sickle Cell Disease In hemoglobin S, a substitution of T for A in the sixth codon of the ßglobin gene leads to the replacement of a glutamic acid residue by a valine residue. On deoxygenation, hemoglobin S polymers form, causing cell sickling and damage to the membrane. Some sickle cells adhere to endothelial cells, leading to vasoocclusion.
 Desaturation of Normal and Variant Hemoglobins during Passage from Artery to Vein Hemoglobins lose oxygen during the journey from artery to vein. Cells with Hb. S sickle only when desaturated. The likelihood that they will obstruct the vessel depends on the concentration of Hb. S and the extent of desaturation.
Desaturation of Normal and Variant Hemoglobins during Passage from Artery to Vein Hemoglobins lose oxygen during the journey from artery to vein. Cells with Hb. S sickle only when desaturated. The likelihood that they will obstruct the vessel depends on the concentration of Hb. S and the extent of desaturation.
 This patient has homozygous sickle cell anemia, presenting with sickle cell ulcers. 1 Leg ulceration is a common complication in patients with homozygous sickle cell anemia. The ulcers appear either spontaneously or as a result of local trauma, and they are chronic. Possible contributing factors are obstruction of vessels by sickled cells, increased venous and capillary pressure, bacterial infection and reduced oxygen-carrying capacity of the blood. 1 These ulcers are common in males, and their incidence increases significantly with advancing age. They occur more often in patients with lower hemoglobin levels and lower fetal hemoglobin levels; patients with SS and SS a-thalassemia have the highest ulcer prevalence rate of all genotypes. These ulcers occur with equal frequency over both medial and lateral aspects of the lower extremities. 1 Treatment modalities include local ulcer care, antibiotics, skin grafting and transfusion therapy. 1 Venous ulcers usually develop above the medial malleoli, in the area drained by incompetent perforating veins. These ulcers are well-demarcated with irregular shaggy borders and surrounding dermatitis, pigmentation and, sometimes, cellulitis.
This patient has homozygous sickle cell anemia, presenting with sickle cell ulcers. 1 Leg ulceration is a common complication in patients with homozygous sickle cell anemia. The ulcers appear either spontaneously or as a result of local trauma, and they are chronic. Possible contributing factors are obstruction of vessels by sickled cells, increased venous and capillary pressure, bacterial infection and reduced oxygen-carrying capacity of the blood. 1 These ulcers are common in males, and their incidence increases significantly with advancing age. They occur more often in patients with lower hemoglobin levels and lower fetal hemoglobin levels; patients with SS and SS a-thalassemia have the highest ulcer prevalence rate of all genotypes. These ulcers occur with equal frequency over both medial and lateral aspects of the lower extremities. 1 Treatment modalities include local ulcer care, antibiotics, skin grafting and transfusion therapy. 1 Venous ulcers usually develop above the medial malleoli, in the area drained by incompetent perforating veins. These ulcers are well-demarcated with irregular shaggy borders and surrounding dermatitis, pigmentation and, sometimes, cellulitis.
 Mechanisms of Action of Hydroxyurea in Sickle Cell Disease
Mechanisms of Action of Hydroxyurea in Sickle Cell Disease




 . Phenotypes of thalassaemias, sickle cell disease and various thalassaemia interactions
. Phenotypes of thalassaemias, sickle cell disease and various thalassaemia interactions
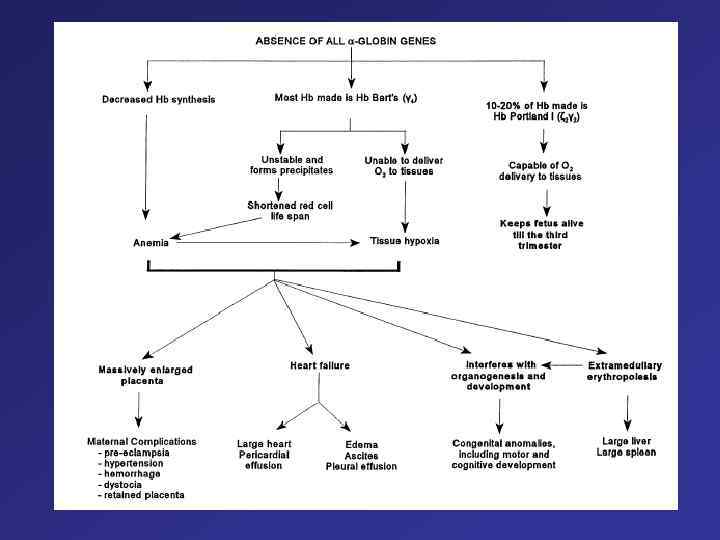
 Clinical and Hematologic Features of the Principal Forms of Thalassemia a-Thalassemias Type of thalassemia Globin-gene expression Hematologic features Clinical expressions Hemoglobin findings a silent carrier - , a/a, a Mild microcytosis or normal Normal a trait - , a/-, a or - , - / a, a Microcytosis, hypochromia, mild anemia Usually normal Newborn: Hb Barts (g 4) 5 to 10 percent Child or adult: normal Hb. H - , a/- , - Microcytosis, inclusion bodies by supravital staining; moderately severe anemia Thalassemia intermedia Newborn: Hb Barts (g 4) 20 to 30 percent Child or adult: Hb. H (b 4) 4 to 20 percent Anisocytosis, poikilocytosis; severe anemia Hydrops fetalis; usually stillborn or neonatal death Hb Barts (g 4), 80 to 90 percent; no Hb. A or Hb. F disease ahydrops fetalis - , -/- , -
Clinical and Hematologic Features of the Principal Forms of Thalassemia a-Thalassemias Type of thalassemia Globin-gene expression Hematologic features Clinical expressions Hemoglobin findings a silent carrier - , a/a, a Mild microcytosis or normal Normal a trait - , a/-, a or - , - / a, a Microcytosis, hypochromia, mild anemia Usually normal Newborn: Hb Barts (g 4) 5 to 10 percent Child or adult: normal Hb. H - , a/- , - Microcytosis, inclusion bodies by supravital staining; moderately severe anemia Thalassemia intermedia Newborn: Hb Barts (g 4) 20 to 30 percent Child or adult: Hb. H (b 4) 4 to 20 percent Anisocytosis, poikilocytosis; severe anemia Hydrops fetalis; usually stillborn or neonatal death Hb Barts (g 4), 80 to 90 percent; no Hb. A or Hb. F disease ahydrops fetalis - , -/- , -
 Autopsy photographs of a newborn with hemoglobin Bart’s hydrops fetalis syndrome, delivered by caesarean section at 31 week of gestation. 59 Despite active resuscitation, the infant died within an hour after birth. Note edema of face and abdominal distension (A). The thoracic cavity is filled by the pericardial sac distended by effusion and cardiomegaly; there is massive hepatomegaly (B).
Autopsy photographs of a newborn with hemoglobin Bart’s hydrops fetalis syndrome, delivered by caesarean section at 31 week of gestation. 59 Despite active resuscitation, the infant died within an hour after birth. Note edema of face and abdominal distension (A). The thoracic cavity is filled by the pericardial sac distended by effusion and cardiomegaly; there is massive hepatomegaly (B).
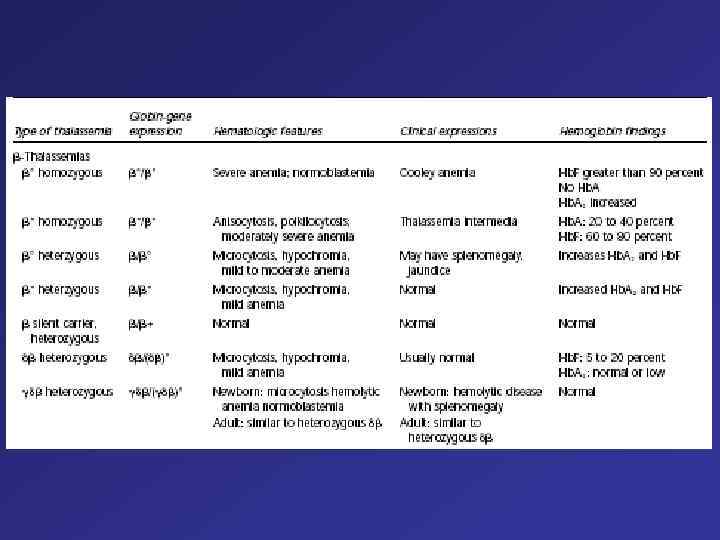
 Clinical and Hematologic Features of the Principal Forms of Thalassemia Type of thalassemia Globin-gene expression Hematologic features Clinical expressions Hemoglobin findings ß° homozygous ß°/ß° Severe anemia; normoblastemia Cooley anemia Hb. F greater than 90 percent No Hb. A 2 increased ß+ homozygous ß+ß+ Anisocytosis, poikilocytosis; moderately severe anemia Thalassemia intermedia Hb. A: 20 to 40 percent Hb. F: 60 to 80 percent ß° heterzygous ß/ß° Microcytosis, hypochromia, mild to moderate anemia May have splenomegaly, jaundice Increases Hb. A 2 and Hb. F ß+ heterzygous ß/ß+ Microcytosis, hypochromia, mild anemia or normal Normal Increased Hb. A 2 and Hb. F or normal
Clinical and Hematologic Features of the Principal Forms of Thalassemia Type of thalassemia Globin-gene expression Hematologic features Clinical expressions Hemoglobin findings ß° homozygous ß°/ß° Severe anemia; normoblastemia Cooley anemia Hb. F greater than 90 percent No Hb. A 2 increased ß+ homozygous ß+ß+ Anisocytosis, poikilocytosis; moderately severe anemia Thalassemia intermedia Hb. A: 20 to 40 percent Hb. F: 60 to 80 percent ß° heterzygous ß/ß° Microcytosis, hypochromia, mild to moderate anemia May have splenomegaly, jaundice Increases Hb. A 2 and Hb. F ß+ heterzygous ß/ß+ Microcytosis, hypochromia, mild anemia or normal Normal Increased Hb. A 2 and Hb. F or normal
 Effects of Excess Production of Free a-Globin Chains Primary disease processes are indicated in orange, and compensatory mechanisms in yellow. Excess unbound -globin chains and their degradation products precipitate in red-cell precursors, causing defective maturation and ineffective erythropoiesis. Hemolysis, owing to the presence of inclusions in the red cells and damage to their membranes by -globin chains and their degradation products, also contributes to the anemia. Anemia stimulates the synthesis of erythropoietin, leading to an intense proliferation of the ineffective marrow, which in turn causes skeletal deformities and a variety of growth and metabolic abnormalities. The anemia is further exacerbated by hemodilution, caused by the shunting of blood through the expanded marrow, and by splenomegaly resulting from entrapment of abnormal red cells in the spleen. Bone marrow expansion also results in characteristic deformities of the skull and face, severe osteopenia, and increased iron absorption.
Effects of Excess Production of Free a-Globin Chains Primary disease processes are indicated in orange, and compensatory mechanisms in yellow. Excess unbound -globin chains and their degradation products precipitate in red-cell precursors, causing defective maturation and ineffective erythropoiesis. Hemolysis, owing to the presence of inclusions in the red cells and damage to their membranes by -globin chains and their degradation products, also contributes to the anemia. Anemia stimulates the synthesis of erythropoietin, leading to an intense proliferation of the ineffective marrow, which in turn causes skeletal deformities and a variety of growth and metabolic abnormalities. The anemia is further exacerbated by hemodilution, caused by the shunting of blood through the expanded marrow, and by splenomegaly resulting from entrapment of abnormal red cells in the spleen. Bone marrow expansion also results in characteristic deformities of the skull and face, severe osteopenia, and increased iron absorption.
 Genetic Sequencing and Protein Analysis of the Patient's Hemoglobin Panel A shows sequencing chromatograms revealing two clone types derived from the patient's DNA. Clone type 1 contained the sickle mutation 20 A T in codon 6 and the spontaneously arising mutation 335 C T in codon 68, indicating that both mutations in the patient were on the same chromosome. Clone type 2 was wild type. Both positions are marked with arrowheads, and the encoded (wild type) amino acids are shown below the chromatograms, with the mutated nucleotide and amino acid underscored. Amplificationrefractory mutation system analysis, shown in Panel B, confirmed the presence of both the wild-type ( C 335) allele and the 335 C T mutation, indicating that the patient was heterozygous for that mutation. Isoelectric focusing of the patient's hemolysate (Panel C) revealed Hb. A, Hb. F, and a band comigrating with Hb. S. Trace Hb. A 2 was also present on the original blot, although it is not visible in the figure. At left is a standard hemoglobin ladder. Hb JP was purified by chromatography on a CM 52 carboxymethylcellulose column (Panel D). The points on the curve represent the absorbance of each collected fraction. The resulting hemoglobin was 93 percent pure on fast protein liquid chromatography (data not shown).
Genetic Sequencing and Protein Analysis of the Patient's Hemoglobin Panel A shows sequencing chromatograms revealing two clone types derived from the patient's DNA. Clone type 1 contained the sickle mutation 20 A T in codon 6 and the spontaneously arising mutation 335 C T in codon 68, indicating that both mutations in the patient were on the same chromosome. Clone type 2 was wild type. Both positions are marked with arrowheads, and the encoded (wild type) amino acids are shown below the chromatograms, with the mutated nucleotide and amino acid underscored. Amplificationrefractory mutation system analysis, shown in Panel B, confirmed the presence of both the wild-type ( C 335) allele and the 335 C T mutation, indicating that the patient was heterozygous for that mutation. Isoelectric focusing of the patient's hemolysate (Panel C) revealed Hb. A, Hb. F, and a band comigrating with Hb. S. Trace Hb. A 2 was also present on the original blot, although it is not visible in the figure. At left is a standard hemoglobin ladder. Hb JP was purified by chromatography on a CM 52 carboxymethylcellulose column (Panel D). The points on the curve represent the absorbance of each collected fraction. The resulting hemoglobin was 93 percent pure on fast protein liquid chromatography (data not shown).
 The Normal Structure of the ß-Globin Gene and the Locations and Types of Mutations Resulting in ß-Thalassemia All ß-globin–like genes contain three exons and two introns between codons 30 and 31 and 104 and 105, respectively. The primary action of all the mutations is to abolish the output of ß-globin chains (ß 0 -thalassemia; shown in red) or reduce the output (ß+-thalassemia; shown in green). The 170 different mutations that act in this way may interfere with the action of the ß-globin gene at the transcriptional level, in the processing of the primary transcript, in the translation of ß-globin messenger RNA, or in the post-translational stability of the ßglobin gene product.
The Normal Structure of the ß-Globin Gene and the Locations and Types of Mutations Resulting in ß-Thalassemia All ß-globin–like genes contain three exons and two introns between codons 30 and 31 and 104 and 105, respectively. The primary action of all the mutations is to abolish the output of ß-globin chains (ß 0 -thalassemia; shown in red) or reduce the output (ß+-thalassemia; shown in green). The 170 different mutations that act in this way may interfere with the action of the ß-globin gene at the transcriptional level, in the processing of the primary transcript, in the translation of ß-globin messenger RNA, or in the post-translational stability of the ßglobin gene product.
 Hepatic Iron Burden over Time and the Effect of Various Hepatic Iron Concentrations in Patients with Thalassemia Major, Homozygous Hemochromatosis, and Heterozygous Hemochromatosis
Hepatic Iron Burden over Time and the Effect of Various Hepatic Iron Concentrations in Patients with Thalassemia Major, Homozygous Hemochromatosis, and Heterozygous Hemochromatosis
 that facilitate vaso-occlusion in sickle") Adhesive interactions between blood and vascular endothelial cells (VEC) that facilitate vaso-occlusion in sickle cell disease. N, neutrophil; M, monocyte; P, platelets; SC, sickle cell; RBC, red blood cell. Double-headed arrows indicate possible adhesion between relevant cells.
Adhesive interactions between blood and vascular endothelial cells (VEC) that facilitate vaso-occlusion in sickle cell disease. N, neutrophil; M, monocyte; P, platelets; SC, sickle cell; RBC, red blood cell. Double-headed arrows indicate possible adhesion between relevant cells.
 The Mode of Inheritance of Forms of Hemoglobin H Disease Involving Deletions and Other Types of Mutations The genotype of the parents is shown at the top. Normal -globin genes are represented by open boxes, and mutated, nondeleted -globin genes ( T) by hatched boxes. The genotypes of the offspring are shown in parentheses.
The Mode of Inheritance of Forms of Hemoglobin H Disease Involving Deletions and Other Types of Mutations The genotype of the parents is shown at the top. Normal -globin genes are represented by open boxes, and mutated, nondeleted -globin genes ( T) by hatched boxes. The genotypes of the offspring are shown in parentheses.
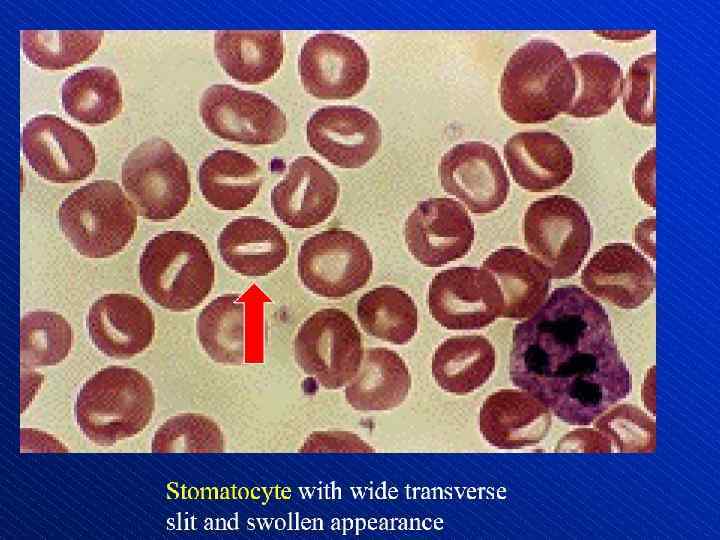
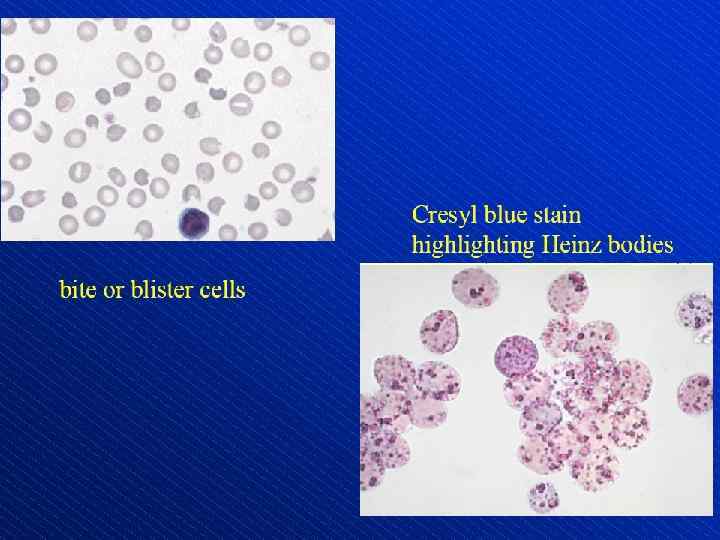
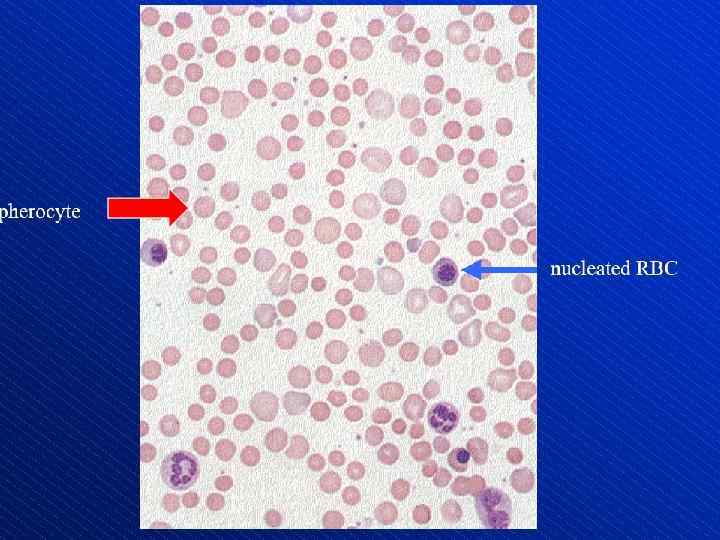
 Depiction of red blood cell morphologies that may appear on a peripheral smear, showing: (A) basophilic stippling, (B) Howell. Jolly bodies, (C) Cabot's ring bodies and (D) Heinz's bodies.
Depiction of red blood cell morphologies that may appear on a peripheral smear, showing: (A) basophilic stippling, (B) Howell. Jolly bodies, (C) Cabot's ring bodies and (D) Heinz's bodies.



 Спасибо за внимание
Спасибо за внимание