

da2eb3593238018a58d1c6783549903e.ppt
- Количество слайдов: 125
 Genome Projects and Gene Hunting Wen-chang Lin Institute of Biomedical Sciences, Academia Sinica Taipei, Taiwan R. O. C. E-mail: wenlin@ibms. sinica. edu. tw Http: //www. ibms. sinica. edu. tw/~wenlin
Genome Projects and Gene Hunting Wen-chang Lin Institute of Biomedical Sciences, Academia Sinica Taipei, Taiwan R. O. C. E-mail: wenlin@ibms. sinica. edu. tw Http: //www. ibms. sinica. edu. tw/~wenlin
 The Human Genome Project is an ambitious effort to understand the hereditary instructions that make each of us unique. The goal of this effort is to find the location of the 100, 000 or so human genes and to read the entire genetic script, all 3 billion bits of information, by the year 2005.
The Human Genome Project is an ambitious effort to understand the hereditary instructions that make each of us unique. The goal of this effort is to find the location of the 100, 000 or so human genes and to read the entire genetic script, all 3 billion bits of information, by the year 2005.
 is an international") What is the Human Genome Project? The Human Genome Project (HGP) is an international research program designed to construct detailed genetic and physical maps of the human genome, to determine the complete nucleotide sequence of human DNA, to localize the estimated 50, 000 -100, 000 genes within the human genome, and to perform similar analyses on the genomes of several other organisms used extensively in research laboratories as model systems. The scientific products of the HGP will comprise a resource of detailed information about the structure, organization and function of human DNA, information that constitutes the basic set of inherited "instructions” for the development and functioning of a human being. Successfully accomplishing these ambitious goals will demand the development of a variety of new technologies. It will also necessitate advanced means of making the information widely available to scientists, physicians, and others in order that the results may be rapidly used for the public good. Improved technology for biomedical research will thus be another important product of the HGP. From the inception of the HGP, it was clearly recognized that acquisition and use of such genetic knowledge would have momentous implications for both individuals and society and would pose a number of policy choices for public and professional deliberation. Analysis of the ethical, legal, and social implications of genetic knowledge, and the development of policy options for public consideration are therefore yet another major component of the human genome research effort.
What is the Human Genome Project? The Human Genome Project (HGP) is an international research program designed to construct detailed genetic and physical maps of the human genome, to determine the complete nucleotide sequence of human DNA, to localize the estimated 50, 000 -100, 000 genes within the human genome, and to perform similar analyses on the genomes of several other organisms used extensively in research laboratories as model systems. The scientific products of the HGP will comprise a resource of detailed information about the structure, organization and function of human DNA, information that constitutes the basic set of inherited "instructions” for the development and functioning of a human being. Successfully accomplishing these ambitious goals will demand the development of a variety of new technologies. It will also necessitate advanced means of making the information widely available to scientists, physicians, and others in order that the results may be rapidly used for the public good. Improved technology for biomedical research will thus be another important product of the HGP. From the inception of the HGP, it was clearly recognized that acquisition and use of such genetic knowledge would have momentous implications for both individuals and society and would pose a number of policy choices for public and professional deliberation. Analysis of the ethical, legal, and social implications of genetic knowledge, and the development of policy options for public consideration are therefore yet another major component of the human genome research effort.
 Genetic Map Complete the 2 -5 c. M") Specific Goals (Phase I 1993 -1998) Genetic Map Complete the 2 -5 c. M map by 1995 Develop technology for rapid genotyping Develop markers that are easier to use Develop new mapping technologies Physical Map Complete an STS map of the human genome at a resolution of 100 kb DNA Sequencing Develop efficient approaches to sequencing one- to several- megabase regions of DNA of high biological interest. Develop technology for high throughput sequencing, focusing on systems integration of all steps from template preparation to data analysis. Build up sequencing capacity to a collective rate of 50 Mb per year by the end of the period. This rate should result in an aggregate of 80 Mb of DNA sequence completed by the end of FY 1998.
Specific Goals (Phase I 1993 -1998) Genetic Map Complete the 2 -5 c. M map by 1995 Develop technology for rapid genotyping Develop markers that are easier to use Develop new mapping technologies Physical Map Complete an STS map of the human genome at a resolution of 100 kb DNA Sequencing Develop efficient approaches to sequencing one- to several- megabase regions of DNA of high biological interest. Develop technology for high throughput sequencing, focusing on systems integration of all steps from template preparation to data analysis. Build up sequencing capacity to a collective rate of 50 Mb per year by the end of the period. This rate should result in an aggregate of 80 Mb of DNA sequence completed by the end of FY 1998.
 Gene Identification Develop efficient methods of identifying genes") Specific Goals (Phase I 1993 -1998) Gene Identification Develop efficient methods of identifying genes and for placement of known genes on physical maps or sequenced DNA. Technology Development Substantially expand support of innovative technological developments as well as improvements in current technology for DNA sequencing and to meet the needs of the Human Genome Project as a whole. Model Organisms Finish an STS map of the mouse at 300 Kb resolution Finish the sequence of the E. coli and S. cerevisiae genomes by 1998 or earlier Continue sequencing C. elegans and Drosophila genomes, with the aim of bringing C. elegans to near completion by 1998 Sequence selected segments of mouse DNA side by side with corresponding human DNA in areas of high biological interest
Specific Goals (Phase I 1993 -1998) Gene Identification Develop efficient methods of identifying genes and for placement of known genes on physical maps or sequenced DNA. Technology Development Substantially expand support of innovative technological developments as well as improvements in current technology for DNA sequencing and to meet the needs of the Human Genome Project as a whole. Model Organisms Finish an STS map of the mouse at 300 Kb resolution Finish the sequence of the E. coli and S. cerevisiae genomes by 1998 or earlier Continue sequencing C. elegans and Drosophila genomes, with the aim of bringing C. elegans to near completion by 1998 Sequence selected segments of mouse DNA side by side with corresponding human DNA in areas of high biological interest
 Informatics Continue to create, develop and operate databases") Specific Goals (Phase I 1993 -1998) Informatics Continue to create, develop and operate databases and database tools for easy access to data, including effective tools and standards for data exchange and links among databases Consolidate, distribute and continue to develop effective software for large-scale genome projects Continue to develop tools for comparing and interpreting genome information Ethical, Legal and Social Implications (ELSI) Continue to identify and define issues and develop policy options to address them Develop and disseminate policy options regarding genetic testing services with widespread potential use Foster greater acceptance of human genetic variation Enhance and expand public and professional education that is sensitive to sociocultural and psychological issues Training Continue to encourage training of scientists in interdisciplinary sciences related to genome research
Specific Goals (Phase I 1993 -1998) Informatics Continue to create, develop and operate databases and database tools for easy access to data, including effective tools and standards for data exchange and links among databases Consolidate, distribute and continue to develop effective software for large-scale genome projects Continue to develop tools for comparing and interpreting genome information Ethical, Legal and Social Implications (ELSI) Continue to identify and define issues and develop policy options to address them Develop and disseminate policy options regarding genetic testing services with widespread potential use Foster greater acceptance of human genetic variation Enhance and expand public and professional education that is sensitive to sociocultural and psychological issues Training Continue to encourage training of scientists in interdisciplinary sciences related to genome research
 Technology Transfer Encourage and enhance technology transfer both") Specific Goals (Phase I 1993 -1998) Technology Transfer Encourage and enhance technology transfer both into and out of centers of genome research Outreach Cooperate with those who would set up distribution centers for genome materials. Share all information and materials within 6 months of their development. This should be accomplished by submission to public databases or repositories, or both, where appropriate.
Specific Goals (Phase I 1993 -1998) Technology Transfer Encourage and enhance technology transfer both into and out of centers of genome research Outreach Cooperate with those who would set up distribution centers for genome materials. Share all information and materials within 6 months of their development. This should be accomplished by submission to public databases or repositories, or both, where appropriate.
") Specific Goals (Phase II 1998 -2003)
Specific Goals (Phase II 1998 -2003)
 Goal 1 --The Human DNA Sequence a) Finish") Specific Goals (Phase II 1998 -2003) Goal 1 --The Human DNA Sequence a) Finish the complete human genome sequence by the end of 2003. b) Finish one-third of the human DNA sequence by the end of 2001. c) Achieve coverage of at least 90% of the genome in a working draft based on mapped clones by the end of 2001. d) Make the sequence totally and freely accessible.
Specific Goals (Phase II 1998 -2003) Goal 1 --The Human DNA Sequence a) Finish the complete human genome sequence by the end of 2003. b) Finish one-third of the human DNA sequence by the end of 2001. c) Achieve coverage of at least 90% of the genome in a working draft based on mapped clones by the end of 2001. d) Make the sequence totally and freely accessible.
 Goal 2 --Sequencing Technology a) Continue to increase") Specific Goals (Phase II 1998 -2003) Goal 2 --Sequencing Technology a) Continue to increase throughput and reduce the cost of current sequencing technology. b) Support research on novel technologies that can lead to significant improvements in sequencing technology. c) Develop effective methods for the advanced development and introduction of new sequencing technologies into the sequencing process.
Specific Goals (Phase II 1998 -2003) Goal 2 --Sequencing Technology a) Continue to increase throughput and reduce the cost of current sequencing technology. b) Support research on novel technologies that can lead to significant improvements in sequencing technology. c) Develop effective methods for the advanced development and introduction of new sequencing technologies into the sequencing process.
 Goal 3 --Human Genome Sequence Variation a) Develop") Specific Goals (Phase II 1998 -2003) Goal 3 --Human Genome Sequence Variation a) Develop technologies for rapid, large-scale identification or scoring, or both, of SNPs and other DNA sequence variants. b) Identify common variants in the coding regions of the majority of identified genes during this 5 -year period. c) Create an SNP map of at least 100, 000 markers. d) Develop the intellectual foundations for studies of sequence variation. e) Create public resources of DNA samples and cell lines.
Specific Goals (Phase II 1998 -2003) Goal 3 --Human Genome Sequence Variation a) Develop technologies for rapid, large-scale identification or scoring, or both, of SNPs and other DNA sequence variants. b) Identify common variants in the coding regions of the majority of identified genes during this 5 -year period. c) Create an SNP map of at least 100, 000 markers. d) Develop the intellectual foundations for studies of sequence variation. e) Create public resources of DNA samples and cell lines.
 Goal 4 --Technology for Functional Genomics a) Develop") Specific Goals (Phase II 1998 -2003) Goal 4 --Technology for Functional Genomics a) Develop c. DNA resources. b) Support research on methods for studying functions of non-protein-coding sequences. c) Develop technology for comprehensive analysis of gene expression. d) Improve methods for genome-wide mutagenesis. e) Develop technology for global protein analysis.
Specific Goals (Phase II 1998 -2003) Goal 4 --Technology for Functional Genomics a) Develop c. DNA resources. b) Support research on methods for studying functions of non-protein-coding sequences. c) Develop technology for comprehensive analysis of gene expression. d) Improve methods for genome-wide mutagenesis. e) Develop technology for global protein analysis.
 Goal 5 --Comparative Genomics a) Complete the sequence") Specific Goals (Phase II 1998 -2003) Goal 5 --Comparative Genomics a) Complete the sequence of the C. elegans genome in 1998. b) Complete the sequence of the Drosophila genome by 2002. c) The mouse genome. 1) Develop physical and genetic mapping resources. 2) Develop additional c. DNA resources. 3) Complete the sequence of the mouse genome by 2005. d) Identify other model organisms that can make major contributions to the understanding of the human genome and support appropriate genomic studies.
Specific Goals (Phase II 1998 -2003) Goal 5 --Comparative Genomics a) Complete the sequence of the C. elegans genome in 1998. b) Complete the sequence of the Drosophila genome by 2002. c) The mouse genome. 1) Develop physical and genetic mapping resources. 2) Develop additional c. DNA resources. 3) Complete the sequence of the mouse genome by 2005. d) Identify other model organisms that can make major contributions to the understanding of the human genome and support appropriate genomic studies.
") Goal 6 --Ethical, Legal, and Social Implications (ELSI)
Goal 6 --Ethical, Legal, and Social Implications (ELSI)
 FY DOE NIH* U. S. Total 1988 10.") U. S. Human Genome Project Funding($Millions) FY DOE NIH* U. S. Total 1988 10. 7 17. 2 27. 9 1989 18. 5 28. 2 46. 7 1990 27. 2 59. 5 86. 7 1991 47. 4 87. 4 134. 8 1992 59. 4 104. 8 164. 2 1993 63. 0 106. 1 169. 1 1994 63. 3 127. 0 190. 3 1995 68. 7 153. 8 222. 5 1996 73. 9 169. 3 243. 2 1997 77. 9 188. 9 266. 8 1998 85. 5 217. 7 303. 2 (NT$9, 780, 500, 000) 1999 89. 8 225. 7 315. 5
U. S. Human Genome Project Funding($Millions) FY DOE NIH* U. S. Total 1988 10. 7 17. 2 27. 9 1989 18. 5 28. 2 46. 7 1990 27. 2 59. 5 86. 7 1991 47. 4 87. 4 134. 8 1992 59. 4 104. 8 164. 2 1993 63. 0 106. 1 169. 1 1994 63. 3 127. 0 190. 3 1995 68. 7 153. 8 222. 5 1996 73. 9 169. 3 243. 2 1997 77. 9 188. 9 266. 8 1998 85. 5 217. 7 303. 2 (NT$9, 780, 500, 000) 1999 89. 8 225. 7 315. 5
 Mar. 24, 2000 -Finished sequence: 561, 973 kb 17. 5% of genome Draft sequence: 2, 020, 129 kb 62. 9% of genome
Mar. 24, 2000 -Finished sequence: 561, 973 kb 17. 5% of genome Draft sequence: 2, 020, 129 kb 62. 9% of genome
 Current Progress Breakdown by Chromosome Chr 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X Y total Effective size (kb) Sequence done (kb) Percent finished Number of contigs Longest contig (kb) 263000 255000 214000 203000 194000 183000 171000 155000 144000 143000 98000 93000 89000 98000 92000 85000 67000 72000 39000 34491 164000 35000 3180491 26571 23193 10417 12521 15679 45668 81476 8730 4839 6091 8398 24509 2143 29775 2196 19372 28861 3734 15021 25825 25851 33620 65513 6934 528043 10. 1% 9. 1% 4. 9% 6. 2% 8. 1% 25. 0% 47. 6% 5. 6% 3. 3% 4. 2% 5. 8% 17. 1% 2. 2% 32. 0% 2. 5% 19. 8% 31. 4% 4. 4% 22. 4% 35. 9% 66. 3% 97. 5% 39. 9% 19. 8% 16. 6 154 109 59 99 94 305 298 42 30 36 63 99 7 106 17 118 129 20 144 137 72 12 347 27 2532 928 695 746 393 739 3926 2094 1902 1010 469 817 1526 1416 1450 297 512 1101 349 1008 1187 7223 23051 949 1104
Current Progress Breakdown by Chromosome Chr 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X Y total Effective size (kb) Sequence done (kb) Percent finished Number of contigs Longest contig (kb) 263000 255000 214000 203000 194000 183000 171000 155000 144000 143000 98000 93000 89000 98000 92000 85000 67000 72000 39000 34491 164000 35000 3180491 26571 23193 10417 12521 15679 45668 81476 8730 4839 6091 8398 24509 2143 29775 2196 19372 28861 3734 15021 25825 25851 33620 65513 6934 528043 10. 1% 9. 1% 4. 9% 6. 2% 8. 1% 25. 0% 47. 6% 5. 6% 3. 3% 4. 2% 5. 8% 17. 1% 2. 2% 32. 0% 2. 5% 19. 8% 31. 4% 4. 4% 22. 4% 35. 9% 66. 3% 97. 5% 39. 9% 19. 8% 16. 6 154 109 59 99 94 305 298 42 30 36 63 99 7 106 17 118 129 20 144 137 72 12 347 27 2532 928 695 746 393 739 3926 2094 1902 1010 469 817 1526 1416 1450 297 512 1101 349 1008 1187 7223 23051 949 1104
 The completed sequence covers 33. 4 Mb of 22 q with 11 gaps and has been estimated to be accurate to less than 1 error in 50, 000 bases, by internal and external checking exercises. The largest contiguous segment stretches over 23 Mb. From our gap-size estimates, we calculate that we have completed 33, 464 kb of a total region spanning 34, 491 kb and that therefore the sequence is complete to 97% coverage of 22 q.
The completed sequence covers 33. 4 Mb of 22 q with 11 gaps and has been estimated to be accurate to less than 1 error in 50, 000 bases, by internal and external checking exercises. The largest contiguous segment stretches over 23 Mb. From our gap-size estimates, we calculate that we have completed 33, 464 kb of a total region spanning 34, 491 kb and that therefore the sequence is complete to 97% coverage of 22 q.
 545 genes; 134 pseudo genes.
545 genes; 134 pseudo genes.
 http: //www. ornl. gov/hgmis/
http: //www. ornl. gov/hgmis/
 3, 000 ~ 4, 000 genes http: //www. ncbi. nlm. nih. gov/disease/
3, 000 ~ 4, 000 genes http: //www. ncbi. nlm. nih. gov/disease/
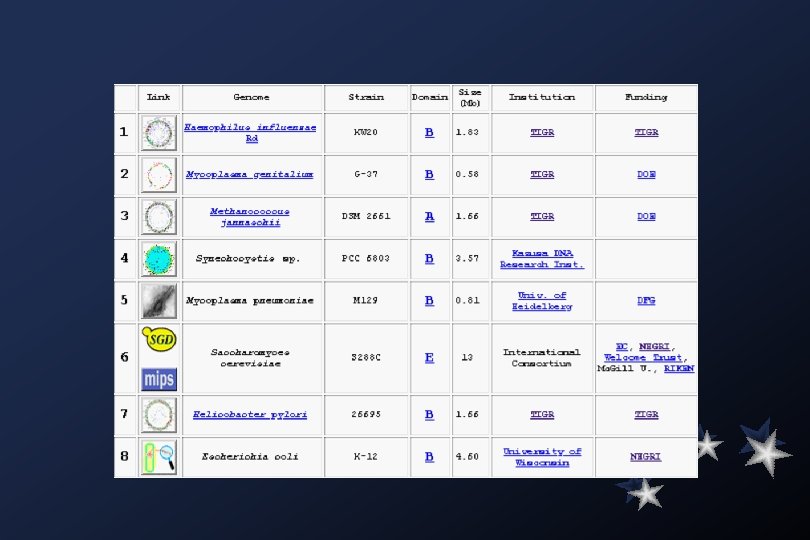
 Caenorhabditis elegans Saccharomyces cerevisiae Escherichia coli Bacillus subtilus") Completed Genomes Organism Genome Size (Mb) Caenorhabditis elegans Saccharomyces cerevisiae Escherichia coli Bacillus subtilus Synechocystis sp. *Archaeoglobus fulgidus *Pyrobaculum aerophilum Haemophilus influenzae *Methanobacterium thermoautotrophicum Helicobacter pylori *Methanococcus jannaschii *Aquifex aolicus Borrelia burgdorferi Treponema pallidum Mycoplasma pneumoniae *Mycoplasma genitalium Treponema pallidum Chlamydia trachomatis Plasmodium falciparum Chr 2 Rickettsia prowazekii Helicobacter pylori Leishmania major Chr 1 100 12. 1 4. 6 4. 2 3. 6 2. 2 1. 8 1. 7 1. 5 1. 3 1. 1 0. 8 0. 6 1. 14 1. 05 1 1. 64. 27 Estimated Genes 6034 4288 ~4000 3168 2471 N. A. 1740 1855 1590 1692 1508 863 1234 677 470
Completed Genomes Organism Genome Size (Mb) Caenorhabditis elegans Saccharomyces cerevisiae Escherichia coli Bacillus subtilus Synechocystis sp. *Archaeoglobus fulgidus *Pyrobaculum aerophilum Haemophilus influenzae *Methanobacterium thermoautotrophicum Helicobacter pylori *Methanococcus jannaschii *Aquifex aolicus Borrelia burgdorferi Treponema pallidum Mycoplasma pneumoniae *Mycoplasma genitalium Treponema pallidum Chlamydia trachomatis Plasmodium falciparum Chr 2 Rickettsia prowazekii Helicobacter pylori Leishmania major Chr 1 100 12. 1 4. 6 4. 2 3. 6 2. 2 1. 8 1. 7 1. 5 1. 3 1. 1 0. 8 0. 6 1. 14 1. 05 1 1. 64. 27 Estimated Genes 6034 4288 ~4000 3168 2471 N. A. 1740 1855 1590 1692 1508 863 1234 677 470
 The TIGR Microbial Database provides links to world-wide genome sequencing projects completed and underway, including the completed TIGR genomes: Archaeoglobus fulgidus, Borreliaburgdorferi, Deinococcus radiodurans, Haemophilus influenzae, Helicobacter pylori, Methanococcus jannaschii, Mycobacterium tuberculosis, Mycoplasma genitalium, Thermotoga maritima, and Treponema pallidum.
The TIGR Microbial Database provides links to world-wide genome sequencing projects completed and underway, including the completed TIGR genomes: Archaeoglobus fulgidus, Borreliaburgdorferi, Deinococcus radiodurans, Haemophilus influenzae, Helicobacter pylori, Methanococcus jannaschii, Mycobacterium tuberculosis, Mycoplasma genitalium, Thermotoga maritima, and Treponema pallidum.
 In the last few decades, advances in molecular biology and the equipment available for research in this field have allowed the increasingly rapid sequencing of large portions of the genomes of several species. In fact, to date, several bacterial genomes, as well as those of some simple eukaryotes (e. g. , Saccharomyces cerevisiae, or baker's yeast) have been sequenced in full. The Human Genome Project, designed to sequence all 24 of the human chromosomes, is also progressing. Popular sequence databases, such as Gen. Bank and EMBL, have been growing at exponential rates. This deluge of information has necessitated the careful storage, organization and indexing of sequence information. Information science has been applied to biology to produce the field called Bioinformatics
In the last few decades, advances in molecular biology and the equipment available for research in this field have allowed the increasingly rapid sequencing of large portions of the genomes of several species. In fact, to date, several bacterial genomes, as well as those of some simple eukaryotes (e. g. , Saccharomyces cerevisiae, or baker's yeast) have been sequenced in full. The Human Genome Project, designed to sequence all 24 of the human chromosomes, is also progressing. Popular sequence databases, such as Gen. Bank and EMBL, have been growing at exponential rates. This deluge of information has necessitated the careful storage, organization and indexing of sequence information. Information science has been applied to biology to produce the field called Bioinformatics
 The most pressing tasks in bioinformatics involve the analysis of sequence information. Computational Biology is the name given to this process, and it involves the following: • • Finding the genes in the DNA sequences of various organisms Developing methods to predict the structure and/or function of newly discovered proteins and structural RNA sequences. Clustering protein sequences into families of related sequences and the development of protein models. Aligning similar proteins and generating phylogenetic trees to examine evolutionary relationships.
The most pressing tasks in bioinformatics involve the analysis of sequence information. Computational Biology is the name given to this process, and it involves the following: • • Finding the genes in the DNA sequences of various organisms Developing methods to predict the structure and/or function of newly discovered proteins and structural RNA sequences. Clustering protein sequences into families of related sequences and the development of protein models. Aligning similar proteins and generating phylogenetic trees to examine evolutionary relationships.
 Simple Mathematics: Human Genome 3 x 10 9 bps Human Genes (5% of the genome) 100, 000 genes In a given cell type at a certain stage, it is estimated that around 20 % of the genes are transcribed or expressed. 20, 000 genes
Simple Mathematics: Human Genome 3 x 10 9 bps Human Genes (5% of the genome) 100, 000 genes In a given cell type at a certain stage, it is estimated that around 20 % of the genes are transcribed or expressed. 20, 000 genes
 Automatic sequencer
Automatic sequencer
 The Growth of Gen. Bank sequence database in the past 10 years. Release Year Base pairs Entries 58 62 66 70 74 80 86 92 98 104 110 115 88 89 90 91 92 93 94 95 96 97 98 99 24, 690, 876 37, 183, 950 51, 306, 092 77, 337, 678 120, 242, 234 163, 802, 597 230, 485, 928 425, 860, 958 730, 552, 938 1, 258, 290, 513 2, 162, 067, 871 4, 653, 932, 745 21, 248 31, 229 41, 057 58, 952 97, 084 150, 744 237, 775 620, 765 1, 114, 581 1, 891, 953 3, 043, 729 5, 354, 511
The Growth of Gen. Bank sequence database in the past 10 years. Release Year Base pairs Entries 58 62 66 70 74 80 86 92 98 104 110 115 88 89 90 91 92 93 94 95 96 97 98 99 24, 690, 876 37, 183, 950 51, 306, 092 77, 337, 678 120, 242, 234 163, 802, 597 230, 485, 928 425, 860, 958 730, 552, 938 1, 258, 290, 513 2, 162, 067, 871 4, 653, 932, 745 21, 248 31, 229 41, 057 58, 952 97, 084 150, 744 237, 775 620, 765 1, 114, 581 1, 891, 953 3, 043, 729 5, 354, 511
 Gene Expression Studies
Gene Expression Studies
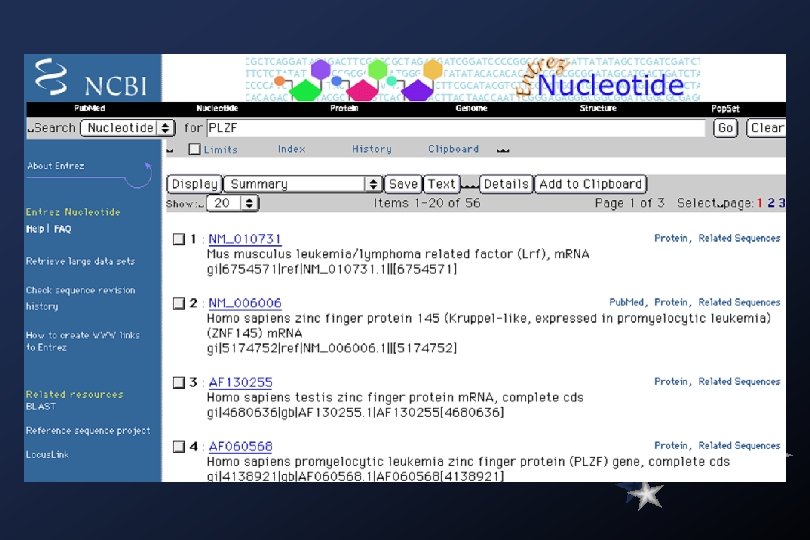
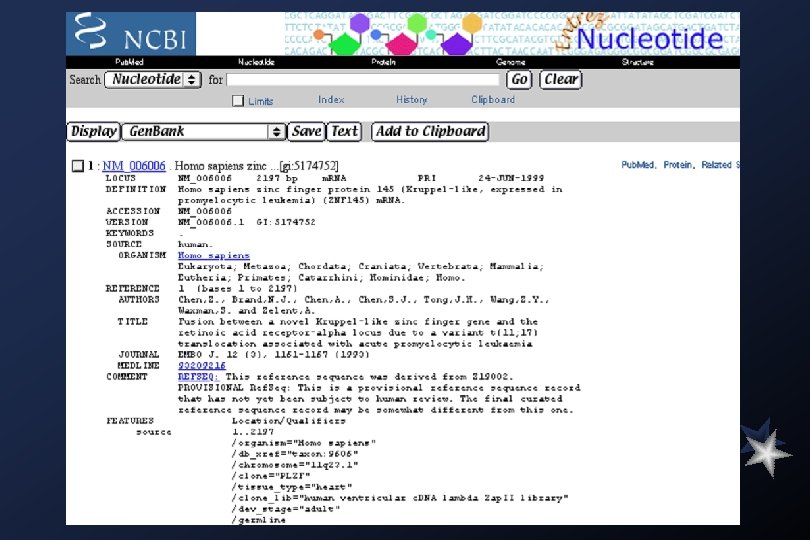
 Gen. Bank Overview What is Gen. Bank? Gen. Bank is the NIH genetic sequence database, an annotated collection of all publicly available DNA sequences ( Nucleic Acids Research 1998 Jan 1; 26(1): 1 -7). There approximately 2, 162, 000 bases in 3, 044, 000 sequence records as of December 1998. As an example, you may view the record for the neurofibromatosis gene. The complete release notes for the current version of Gen. Bank are available. A new release is made every two months. Gen. Bank is part of the International Nucleotide Sequence Database Collaboration, which is comprised of the DNA Data. Bank of Japan (DDBJ), the European Molecular Biology Laboratory (EMBL), and Gen. Bank at NCBI. These three organizations exchange data on a daily basis. Submissions to Gen. Bank Many journals require submission of sequence information to a database prior to publication so that an accession number may appear in the paper. NCBI has a WWW form, called Bank. It, for convenient and quick submission of sequence data. The beta-test version of Sequin, NCBI's new stand-alone submission software for MAC, PC, and UNIX platforms, is available by FTP. When using Sequin, the output files for direct submission should be sent to Gen. Bank by electronic mail. Alternatively, the data files may be copied to a floppy disk and mailed to NCBI. Authorin, an older stand-alone program for MACs and PCs, can still be used to format your submission, although submitters are encouraged to switch to either Bank. It or Sequin.
Gen. Bank Overview What is Gen. Bank? Gen. Bank is the NIH genetic sequence database, an annotated collection of all publicly available DNA sequences ( Nucleic Acids Research 1998 Jan 1; 26(1): 1 -7). There approximately 2, 162, 000 bases in 3, 044, 000 sequence records as of December 1998. As an example, you may view the record for the neurofibromatosis gene. The complete release notes for the current version of Gen. Bank are available. A new release is made every two months. Gen. Bank is part of the International Nucleotide Sequence Database Collaboration, which is comprised of the DNA Data. Bank of Japan (DDBJ), the European Molecular Biology Laboratory (EMBL), and Gen. Bank at NCBI. These three organizations exchange data on a daily basis. Submissions to Gen. Bank Many journals require submission of sequence information to a database prior to publication so that an accession number may appear in the paper. NCBI has a WWW form, called Bank. It, for convenient and quick submission of sequence data. The beta-test version of Sequin, NCBI's new stand-alone submission software for MAC, PC, and UNIX platforms, is available by FTP. When using Sequin, the output files for direct submission should be sent to Gen. Bank by electronic mail. Alternatively, the data files may be copied to a floppy disk and mailed to NCBI. Authorin, an older stand-alone program for MACs and PCs, can still be used to format your submission, although submitters are encouraged to switch to either Bank. It or Sequin.
,") Searching Gen. Bank Text and Similarity searching Entrez Browser Gen. Bank (nucleotides and proteins), Pub. Med (MEDLINE), 3 D structures, genomes, and taxonomy databases. BLAST Sequence Similarity Searching Nucleotide or protein query sequences against the specified database using the BLAST suite of algorithms. db. EST Searching db. EST (Database of Expressed Sequence Tags).
Searching Gen. Bank Text and Similarity searching Entrez Browser Gen. Bank (nucleotides and proteins), Pub. Med (MEDLINE), 3 D structures, genomes, and taxonomy databases. BLAST Sequence Similarity Searching Nucleotide or protein query sequences against the specified database using the BLAST suite of algorithms. db. EST Searching db. EST (Database of Expressed Sequence Tags).
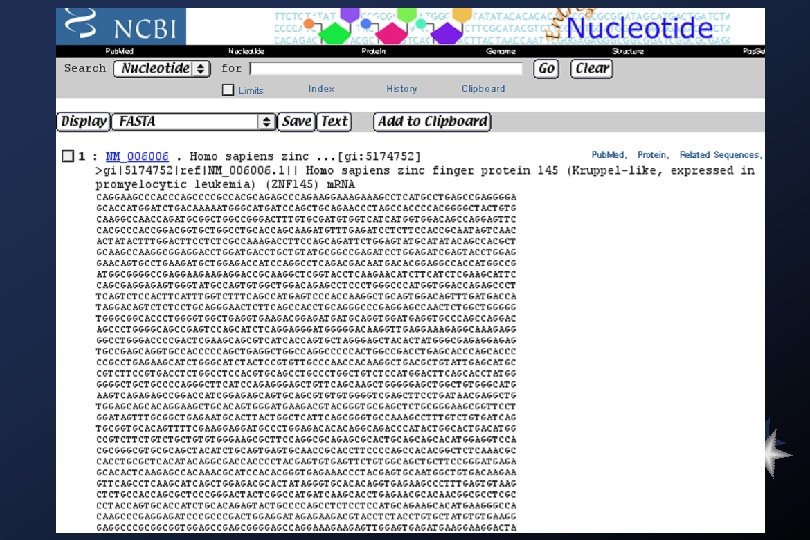
 Gen. Bank nr database: >gi|216185|dbj|D 00635|ABCADHCC Acetobacter polyoxogenes for alcohol dehydrogenase, cytochrome c, complete cds ¶ GAATTCCGAACTATCCGTTTCATTGCTTATGCGACAGCATGTTCACTTTTTAGTGAGGCTGAACACTAAA ATGTCAGGAGACGAGCGTGCTAGCCTCAGTATGTTGCCATGAAACGGACCACCTGCTTTGTCTTTCCTGC CTGAAGCCGGTTTCTGGCCGGAAAAGAAGCGCTAGCGCGTTTTTTTGCCGGATACATTCAGAAAGC TGCTCCGGGCAGAAAGTTGCAGCGGCGGCATCCTGAATTCGAAACCGTTAGTTTTCTGAGGACATCACAT ATGATTTCTGCCGTTTTCGGAAAAAGACGTTCTCTGAGCAGAACGCTTACAGCCGGAACGATATGTGCGG CTCTCATCTCCGGGTATGCCACCATGGCATCCGCAGATGACGGGCAGGGCGCCACGGGGGAAGCGATCAT CCATGCCGATGATCACCCCGGTAACTGGATGACCTATGGCCGCACCTATTCTGACCAGCGCTACAGCCCG CTGGATCAACCGTTCCAATGTCGGTAACCTGAAGCTGGCCTGGTATCTGGACCTTGATACCAACC GTGGCCAGGAAGGCACGCCCCTGGTTATTGATGGCGTCATGTACGCCACCACCAACTGGAGCATGATGAA AGCCGTCGACGCCGCAACCGGCAAGCTGCTGTGGTCCTATGACCCGCGCGTGCCCGGCAACATTGCCGAC AAGGGCTGCTGTGACACGGTCAACCGTGGCGCGGCATACTGGAATGGCAAGGTCTATTTCGGCACGTTCG ACGGTCGCCTGATCGCGCTGGACGCCAAGACCGGCAAGCTGGTCTGGAGCGTCAACACCATTCCGCCCGA AGCGGAACTGGGCAAGCAGCGTTCCTATACGGTTGACGGCGCGCCCCGTATCGCCAAGGGCCGCGTGA>> ¶ FASTA format
Gen. Bank nr database: >gi|216185|dbj|D 00635|ABCADHCC Acetobacter polyoxogenes for alcohol dehydrogenase, cytochrome c, complete cds ¶ GAATTCCGAACTATCCGTTTCATTGCTTATGCGACAGCATGTTCACTTTTTAGTGAGGCTGAACACTAAA ATGTCAGGAGACGAGCGTGCTAGCCTCAGTATGTTGCCATGAAACGGACCACCTGCTTTGTCTTTCCTGC CTGAAGCCGGTTTCTGGCCGGAAAAGAAGCGCTAGCGCGTTTTTTTGCCGGATACATTCAGAAAGC TGCTCCGGGCAGAAAGTTGCAGCGGCGGCATCCTGAATTCGAAACCGTTAGTTTTCTGAGGACATCACAT ATGATTTCTGCCGTTTTCGGAAAAAGACGTTCTCTGAGCAGAACGCTTACAGCCGGAACGATATGTGCGG CTCTCATCTCCGGGTATGCCACCATGGCATCCGCAGATGACGGGCAGGGCGCCACGGGGGAAGCGATCAT CCATGCCGATGATCACCCCGGTAACTGGATGACCTATGGCCGCACCTATTCTGACCAGCGCTACAGCCCG CTGGATCAACCGTTCCAATGTCGGTAACCTGAAGCTGGCCTGGTATCTGGACCTTGATACCAACC GTGGCCAGGAAGGCACGCCCCTGGTTATTGATGGCGTCATGTACGCCACCACCAACTGGAGCATGATGAA AGCCGTCGACGCCGCAACCGGCAAGCTGCTGTGGTCCTATGACCCGCGCGTGCCCGGCAACATTGCCGAC AAGGGCTGCTGTGACACGGTCAACCGTGGCGCGGCATACTGGAATGGCAAGGTCTATTTCGGCACGTTCG ACGGTCGCCTGATCGCGCTGGACGCCAAGACCGGCAAGCTGGTCTGGAGCGTCAACACCATTCCGCCCGA AGCGGAACTGGGCAAGCAGCGTTCCTATACGGTTGACGGCGCGCCCCGTATCGCCAAGGGCCGCGTGA>> ¶ FASTA format





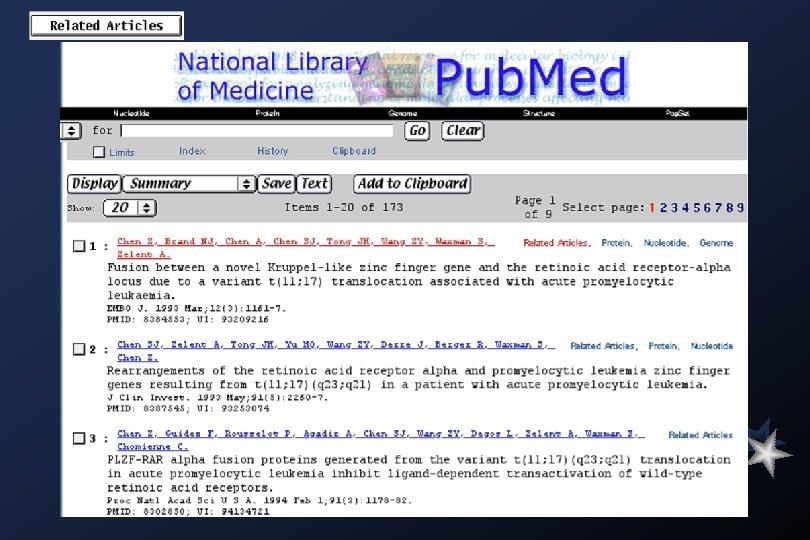
") Medline searches: Academia Sinica Library (local)
Medline searches: Academia Sinica Library (local)
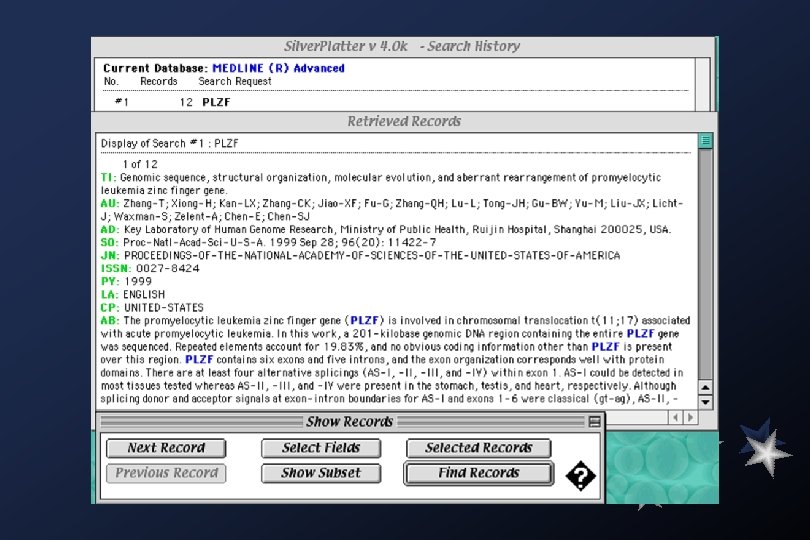
 Http: //igm. nlm. nih. gov/
Http: //igm. nlm. nih. gov/
 Given COX-1 and COX-2 can a putative COX-3 be identified? Text search for COX-3 (and suitable alternative forms) Acquire human COX-1 and COX-2 sequences Search for sequence similarties in a fulllength sequence database Search for sequence similarties in an EST database Merge the results of the full-length and EST searches
Given COX-1 and COX-2 can a putative COX-3 be identified? Text search for COX-3 (and suitable alternative forms) Acquire human COX-1 and COX-2 sequences Search for sequence similarties in a fulllength sequence database Search for sequence similarties in an EST database Merge the results of the full-length and EST searches
 ESTs virtually indentical to COX-1 and COX-2 ESTs similar, but not indentical to COX-1/-2 May provide tissue localization information Search ESTs back against full-length databases Strong similarities with other genes indicate close relationship of COX family to another gene family probably with a different function Is it highly similar to COX -1, COX-2 or both? Is it only weakly similar? If so, might it be more similar to something else, a putative COX-3?
ESTs virtually indentical to COX-1 and COX-2 ESTs similar, but not indentical to COX-1/-2 May provide tissue localization information Search ESTs back against full-length databases Strong similarities with other genes indicate close relationship of COX family to another gene family probably with a different function Is it highly similar to COX -1, COX-2 or both? Is it only weakly similar? If so, might it be more similar to something else, a putative COX-3?
 In silico cloning: In order to perform an electronic c. DNA library screen, the EST sequences retrieved in this way can be used as queries in a BLASTN search of db. EST to identify over-lapping ESTs. This procedure can be reiterated with the newly identified ESTs until no additional hits are found. The ESTs isolated can be assembled into sequence contigs using computer softwares. EST 1 EST 3 Query EST 2
In silico cloning: In order to perform an electronic c. DNA library screen, the EST sequences retrieved in this way can be used as queries in a BLASTN search of db. EST to identify over-lapping ESTs. This procedure can be reiterated with the newly identified ESTs until no additional hits are found. The ESTs isolated can be assembled into sequence contigs using computer softwares. EST 1 EST 3 Query EST 2
 1 61 121 181 241 301 361 421 481 541 601 661 mdltkmgmiq ilfhrnsqhy mletiqasdd vdqspsvsts vktemmqvde esaeqvpppa alavsmdfst qhrklhsgmk qthtgtdmav tgdhpyecef pfecklchqr wriektylyl lqnpshptgl tldflspktf ndteatmadg fglsamsptk vpsqdspgaa eagqaptgrp yggllpqgfi tygcelcgkr fcllcgkrfq cgscfrdest srdysamikh cyv lckanqmrla qqileyayta gaeeeedrka aavdslmtig essisggmgd ehpapppekh qrelfsklge fldslrlrmh aqsalqqhme lkshkrihtg lrthngaspy gtlcdvvimv tlqakaedld rylknifisk qsllqgtlqp kveergkegp lgiysvlpnh lavgmksesr llahsagaka vhagvrsyic ekpyecngcd qcticteycp dsqefhahrt dllyaaeile hsseesgyas pagpeeptla gtptrssvit kadavlsmps tigeqcsvcg fvcdqcgaqf secnrtfpsh kkfslkhqle slssmqkhmk vlactskmfe ieyleeqclk vagqslpgpm gggrhpgvae sarelhygre svtsglhvqp velpdneave skedalethr talkrhlrsh thyrvhtgek ghkpeeippd
1 61 121 181 241 301 361 421 481 541 601 661 mdltkmgmiq ilfhrnsqhy mletiqasdd vdqspsvsts vktemmqvde esaeqvpppa alavsmdfst qhrklhsgmk qthtgtdmav tgdhpyecef pfecklchqr wriektylyl lqnpshptgl tldflspktf ndteatmadg fglsamsptk vpsqdspgaa eagqaptgrp yggllpqgfi tygcelcgkr fcllcgkrfq cgscfrdest srdysamikh cyv lckanqmrla qqileyayta gaeeeedrka aavdslmtig essisggmgd ehpapppekh qrelfsklge fldslrlrmh aqsalqqhme lkshkrihtg lrthngaspy gtlcdvvimv tlqakaedld rylknifisk qsllqgtlqp kveergkegp lgiysvlpnh lavgmksesr llahsagaka vhagvrsyic ekpyecngcd qcticteycp dsqefhahrt dllyaaeile hsseesgyas pagpeeptla gtptrssvit kadavlsmps tigeqcsvcg fvcdqcgaqf secnrtfpsh kkfslkhqle slssmqkhmk vlactskmfe ieyleeqclk vagqslpgpm gggrhpgvae sarelhygre svtsglhvqp velpdneave skedalethr talkrhlrsh thyrvhtgek ghkpeeippd
 Sequence Alignment and Similarity Search: One goal of sequence alignment is to enable the researcher to determine whether two sequences display sufficient similarity to justify the inference of homology. Similarity is an observable quantity that might be expressed as, say, percent identity or some other suitable measure. Homology, on the other hand, refers to a conclusion drawn from these data that two genes share a common evolutionary history. While it is presumed that homologous sequences have diverged from a common ancestral sequence through iterative changes, we do not actually know what the ancestral sequence was (barring the possibility that DNA could be recovered from a fossil); all we have to observe are the sequences from extant organisms. In a residue-by-residue alignment it is often apparent that certain regions of a protein, or perhaps specific amino acids, are more highly conserved than others. This information may be suggestive of which residues are most crucial for a maintaining a protein’s structure or function.
Sequence Alignment and Similarity Search: One goal of sequence alignment is to enable the researcher to determine whether two sequences display sufficient similarity to justify the inference of homology. Similarity is an observable quantity that might be expressed as, say, percent identity or some other suitable measure. Homology, on the other hand, refers to a conclusion drawn from these data that two genes share a common evolutionary history. While it is presumed that homologous sequences have diverged from a common ancestral sequence through iterative changes, we do not actually know what the ancestral sequence was (barring the possibility that DNA could be recovered from a fossil); all we have to observe are the sequences from extant organisms. In a residue-by-residue alignment it is often apparent that certain regions of a protein, or perhaps specific amino acids, are more highly conserved than others. This information may be suggestive of which residues are most crucial for a maintaining a protein’s structure or function.
 hum p. LZF p 1 MDLTKMGMIQLQNPSHPTGLLCKANQMRLAGTLCDVVIMVDSQEFHAHRTVLACTSKMFE 60 hum TZFP p 1 MSLPPIRLPSPYGSDRLVQLAARLRPA--LCDTLITVGSQEFPAHSLVLAGVSQQLG 55 : I: L P L: A : : R A LCD : I V SQEF AH VLA S: hum p. LZF p 61 ILFHRNSQHYTLDFLSPKTFQQILEYAYTATLQAKAEDLDDLLYAAEILEIEYLEEQCLK 120 hum TZFP p 56 ----RRGQWALGEGISPSTFAQLLNFVYGESVELQPGELRPLQEAARALGVQSLEEACWR 111 R Q : : SP TF Q: L : Y : : : L L AA L : : LEE C : hum p. LZF p 121 MLETIQASDDNDTEATMADGGAEEEEDRKARYLKNIFISKHSSEESGYASVAGQSLPGPM 180 hum TZFP p 112 ARGDRAKKPDP----G---------LKKHQEEPEKPSRNPERELGDPG 146 D G : KH E : : L P hum p. LZF p 181 VDQSP-SVSTSFGLSAMSPTKAAVDSLMTIGQSLLQGTLQPPAGPEEPTLAGGGRHPGVA 239 hum TZFP p 147 EKQKPEQVSRTGGR---------EQEMLHKHSPPRG--RPEMAG---- 179 Q P VS : G : : PP G P : AG hum p. LZF p 240 EVKTEMMQVDEVPSQDSPGAAESSISGGMGDKVEERGKEGPGTPTRSSVITSARELHYGR 299 hum TZFP p 180 --ATQEAQQEQTRSK------EKRLQAPVG----QRGADG-----KHGVLTWLRENPGGS 222 T: Q : : S: E : : G : RG : V: T RE G hum p. LZF p 300 EESAEQVPPPAEAGQAPTGRPEHPAPP-PEKHLGIYSVLPNHKADAVLSMPSSVTSGLHV 358 hum TZFP p 223 EESLRKLPGPLP----PAGSLQTSVTPRPSWAEAPWLVGGQPALWSILLMPP---- 270 EES : : P P P: G : P P V : : : L MP
hum p. LZF p 1 MDLTKMGMIQLQNPSHPTGLLCKANQMRLAGTLCDVVIMVDSQEFHAHRTVLACTSKMFE 60 hum TZFP p 1 MSLPPIRLPSPYGSDRLVQLAARLRPA--LCDTLITVGSQEFPAHSLVLAGVSQQLG 55 : I: L P L: A : : R A LCD : I V SQEF AH VLA S: hum p. LZF p 61 ILFHRNSQHYTLDFLSPKTFQQILEYAYTATLQAKAEDLDDLLYAAEILEIEYLEEQCLK 120 hum TZFP p 56 ----RRGQWALGEGISPSTFAQLLNFVYGESVELQPGELRPLQEAARALGVQSLEEACWR 111 R Q : : SP TF Q: L : Y : : : L L AA L : : LEE C : hum p. LZF p 121 MLETIQASDDNDTEATMADGGAEEEEDRKARYLKNIFISKHSSEESGYASVAGQSLPGPM 180 hum TZFP p 112 ARGDRAKKPDP----G---------LKKHQEEPEKPSRNPERELGDPG 146 D G : KH E : : L P hum p. LZF p 181 VDQSP-SVSTSFGLSAMSPTKAAVDSLMTIGQSLLQGTLQPPAGPEEPTLAGGGRHPGVA 239 hum TZFP p 147 EKQKPEQVSRTGGR---------EQEMLHKHSPPRG--RPEMAG---- 179 Q P VS : G : : PP G P : AG hum p. LZF p 240 EVKTEMMQVDEVPSQDSPGAAESSISGGMGDKVEERGKEGPGTPTRSSVITSARELHYGR 299 hum TZFP p 180 --ATQEAQQEQTRSK------EKRLQAPVG----QRGADG-----KHGVLTWLRENPGGS 222 T: Q : : S: E : : G : RG : V: T RE G hum p. LZF p 300 EESAEQVPPPAEAGQAPTGRPEHPAPP-PEKHLGIYSVLPNHKADAVLSMPSSVTSGLHV 358 hum TZFP p 223 EESLRKLPGPLP----PAGSLQTSVTPRPSWAEAPWLVGGQPALWSILLMPP---- 270 EES : : P P P: G : P P V : : : L MP
 hum p. LZF p 359 QPALAVSMDFSTYGGLLPQGFIQRELFSKLGELAVGMKSESRTIGEQCSVCGVELPDNEA 418 hum TZFP p 271 RYGIPFYHSTPTTGAWQEVWREQRIPLSLNAPKGLWSQNQ---L-ASSSPTPGSLP---- 322 : : T G QR S : : : S : LP hum p. LZF p 419 VEQHRKLHSGMKTYGCELCGKRFLDSLRLRMHLLAHSAGAKAFVCDQCGAQFSKEDALET 478 hum TZFP p 323 -----------------------QGPAQLS-PGEMEE 335 Q AQ S : E hum p. LZF p 479 HRQTHTGTDMAVFCLLCGKRFQAQSALQQHMEVHAGVRSYICSECNRTFPSHTALKRHLR 538 hum TZFP p 336 SDQGHTG--------ALATCAGHEDKAG------CPPRPHPPPAPPARSR--- 371 Q HTG A : : H : C : P: A R hum p. LZF p 539 SHTGDHPYECEFCGSCFRDESTLKSHKRIHTGEKPYECNGCDKKFSLKHQLETHYRVHTG 598 hum TZFP p 372 -----------------PYACSVCGKRFSLKHQMETHYRVHTG 397 PY C C K: FSLKHQ: ETHYRVHTG hum p. LZF p 599 EKPFECKLCHQRSRDYSAMIKHLRTHNGASPYQCTICTEYCPSLSSMQKHMKGHKPEEIP 658 hum TZFP p 398 EKPFSCSLCPQRSRDFSAMTKHLRTH-GAAPYRCSLCGAGCPSLASMQAHMRGHSPSQLP 456 EKPF C LC QRSRD: SAM KHLRTH GA: PY: C: : C CPSL: SMQ HM: GH P : : P hum p. LZF p 659 PDWRIEKTYLY------LCYV 673 hum TZFP p 457 PGWTIRSTFLYSSSRPSRPSTSPCCPSSSTT 487 P W I T: LY C
hum p. LZF p 359 QPALAVSMDFSTYGGLLPQGFIQRELFSKLGELAVGMKSESRTIGEQCSVCGVELPDNEA 418 hum TZFP p 271 RYGIPFYHSTPTTGAWQEVWREQRIPLSLNAPKGLWSQNQ---L-ASSSPTPGSLP---- 322 : : T G QR S : : : S : LP hum p. LZF p 419 VEQHRKLHSGMKTYGCELCGKRFLDSLRLRMHLLAHSAGAKAFVCDQCGAQFSKEDALET 478 hum TZFP p 323 -----------------------QGPAQLS-PGEMEE 335 Q AQ S : E hum p. LZF p 479 HRQTHTGTDMAVFCLLCGKRFQAQSALQQHMEVHAGVRSYICSECNRTFPSHTALKRHLR 538 hum TZFP p 336 SDQGHTG--------ALATCAGHEDKAG------CPPRPHPPPAPPARSR--- 371 Q HTG A : : H : C : P: A R hum p. LZF p 539 SHTGDHPYECEFCGSCFRDESTLKSHKRIHTGEKPYECNGCDKKFSLKHQLETHYRVHTG 598 hum TZFP p 372 -----------------PYACSVCGKRFSLKHQMETHYRVHTG 397 PY C C K: FSLKHQ: ETHYRVHTG hum p. LZF p 599 EKPFECKLCHQRSRDYSAMIKHLRTHNGASPYQCTICTEYCPSLSSMQKHMKGHKPEEIP 658 hum TZFP p 398 EKPFSCSLCPQRSRDFSAMTKHLRTH-GAAPYRCSLCGAGCPSLASMQAHMRGHSPSQLP 456 EKPF C LC QRSRD: SAM KHLRTH GA: PY: C: : C CPSL: SMQ HM: GH P : : P hum p. LZF p 659 PDWRIEKTYLY------LCYV 673 hum TZFP p 457 PGWTIRSTFLYSSSRPSRPSTSPCCPSSSTT 487 P W I T: LY C
 Sequence Alignment and Similarity Search: Database similarity searching allows us to determine which of the hundreds of thousands of sequences present in the database are potentially related to a particular sequence of interest. In database searching, the basic operation is to sequentially align a query sequence to each subject sequence in the database. The results are reported as a ranked hit list followed by a series of individual sequence alignments, plus various scores and statistics. Current sequence databases are already immense and have continued to increase at an exponential rate, making straightforward application of dynamic programming methods impractical for database searching. One solution is to use massively parallel computers. There are several frequently used programs available on the Internet: Fast. A BLITZ BLAST Smith-Waterman based system (Gen. Web of NHRI)
Sequence Alignment and Similarity Search: Database similarity searching allows us to determine which of the hundreds of thousands of sequences present in the database are potentially related to a particular sequence of interest. In database searching, the basic operation is to sequentially align a query sequence to each subject sequence in the database. The results are reported as a ranked hit list followed by a series of individual sequence alignments, plus various scores and statistics. Current sequence databases are already immense and have continued to increase at an exponential rate, making straightforward application of dynamic programming methods impractical for database searching. One solution is to use massively parallel computers. There are several frequently used programs available on the Internet: Fast. A BLITZ BLAST Smith-Waterman based system (Gen. Web of NHRI)
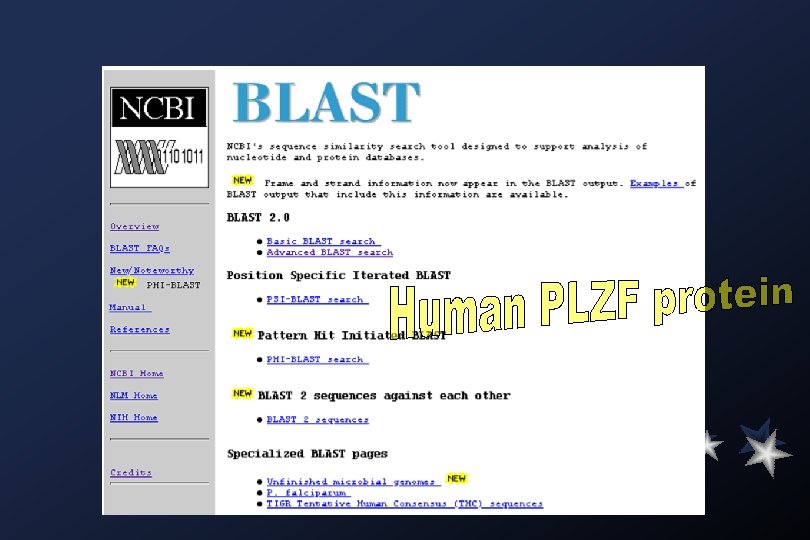
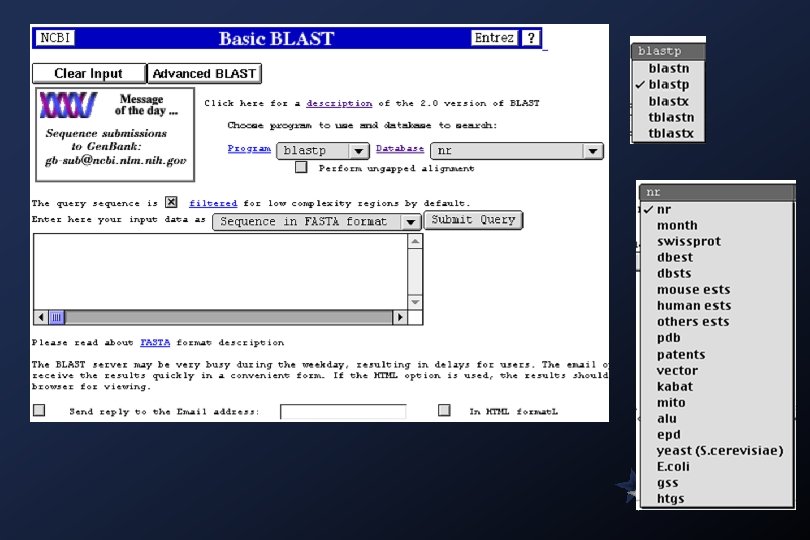
 Blast Family of Programs The BLAST family of programs allows all combinations of DNA or protein query sequences with searches against DNA or protein databases: blastp compares an amino acid query sequence against a protein sequence database. blastn compares a nucleotide query sequence against a nucleotide sequence database. blastx compares the six-frame conceptual translation products of a nucleotide query sequence (both strands) against a protein sequence database. tblastn compares a protein query sequence against a nucleotide sequence database dynamically translated in all six reading frames (both strands). tblastx compares the six-frame translations of a nucleotide query sequence against the six-frame translations of a nucleotide sequence database. The default matrix for all protein-protein comparisons is BLOSUM 62.
Blast Family of Programs The BLAST family of programs allows all combinations of DNA or protein query sequences with searches against DNA or protein databases: blastp compares an amino acid query sequence against a protein sequence database. blastn compares a nucleotide query sequence against a nucleotide sequence database. blastx compares the six-frame conceptual translation products of a nucleotide query sequence (both strands) against a protein sequence database. tblastn compares a protein query sequence against a nucleotide sequence database dynamically translated in all six reading frames (both strands). tblastx compares the six-frame translations of a nucleotide query sequence against the six-frame translations of a nucleotide sequence database. The default matrix for all protein-protein comparisons is BLOSUM 62.
 Databases available for BLAST search Protein Sequence Databases nr All non-redundant Gen. Bank CDS translations+PDB+Swiss. Prot+PIR+PRF month All new or revised Gen. Bank CDS translation+PDB+Swiss. Prot+PIR+PRF released in the last 30 days. swissprot the last major release of the SWISS-PROT protein sequence database (no updates) yeast Yeast (Saccharomyces cerevisiae) protein sequences. E. coli genomic CDS translations pdb Sequences derived from the 3 -dimensional structure Brookhaven Protein Data Bank Nucleotide Sequence Databases nr All Non-redundant Gen. Bank+EMBL+DDBJ+PDB sequences (but no EST, STS, GSS, or HTGS sequences) month All new or revised Gen. Bank+EMBL+DDBJ+PDB sequences released in the last 30 days. dbest Non-redundant Database of Gen. Bank+EMBL+DDBJ EST Divisions dbsts Non-redundant Database of Gen. Bank+EMBL+DDBJ STS Divisions yeast Yeast (Saccharomyces cerevisiae) genomic nucleotide sequences E. coli genomic nucleotide sequences
Databases available for BLAST search Protein Sequence Databases nr All non-redundant Gen. Bank CDS translations+PDB+Swiss. Prot+PIR+PRF month All new or revised Gen. Bank CDS translation+PDB+Swiss. Prot+PIR+PRF released in the last 30 days. swissprot the last major release of the SWISS-PROT protein sequence database (no updates) yeast Yeast (Saccharomyces cerevisiae) protein sequences. E. coli genomic CDS translations pdb Sequences derived from the 3 -dimensional structure Brookhaven Protein Data Bank Nucleotide Sequence Databases nr All Non-redundant Gen. Bank+EMBL+DDBJ+PDB sequences (but no EST, STS, GSS, or HTGS sequences) month All new or revised Gen. Bank+EMBL+DDBJ+PDB sequences released in the last 30 days. dbest Non-redundant Database of Gen. Bank+EMBL+DDBJ EST Divisions dbsts Non-redundant Database of Gen. Bank+EMBL+DDBJ STS Divisions yeast Yeast (Saccharomyces cerevisiae) genomic nucleotide sequences E. coli genomic nucleotide sequences


 organism
organism

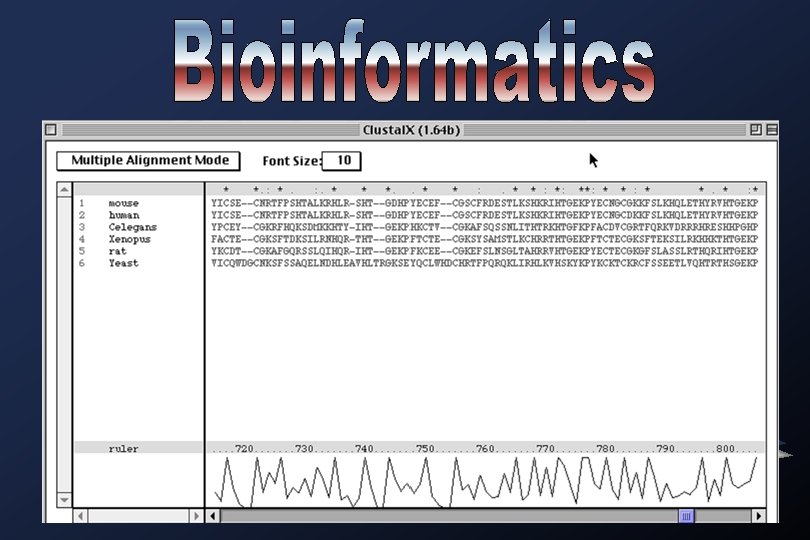
 CLUSTAL W One of the most widely used multiple sequence alignment program. Based on the idea of progressive alignment, this program takes an input set of sequences and calculates a series of pairwise alignments, comparing each sequence to every other sequence, one at a time.
CLUSTAL W One of the most widely used multiple sequence alignment program. Based on the idea of progressive alignment, this program takes an input set of sequences and calculates a series of pairwise alignments, comparing each sequence to every other sequence, one at a time.
 (part #2) (part #3) 406 409 426") Human PLZF 406 ZINC 1 (part #1) (part #2) (part #3) 406 409 426 1/1 14/14 1/1 C CGVELPDNEAVEQH H (part #1) (part #2) (part #3) 434 437 454 1/1 14/14 1/1 C CGKRFLDSLRLRMH H (part #1) (part #2) (part #3) 463 466 483 1/1 14/14 1/1 C CGAQFSKEDALETH H (part #1) (part #2) (part #3) 492 495 512 1/1 14/14 1/1 C CGKRFQAQSALQQH H (part #1) (part #2) (part #3) 520 523 540 1/1 14/14 1/1 C CNRTFPSHTALKRH H 434 ZINC 1 463 ZINC 1 492 ZINC 1 520 ZINC 1
Human PLZF 406 ZINC 1 (part #1) (part #2) (part #3) 406 409 426 1/1 14/14 1/1 C CGVELPDNEAVEQH H (part #1) (part #2) (part #3) 434 437 454 1/1 14/14 1/1 C CGKRFLDSLRLRMH H (part #1) (part #2) (part #3) 463 466 483 1/1 14/14 1/1 C CGAQFSKEDALETH H (part #1) (part #2) (part #3) 492 495 512 1/1 14/14 1/1 C CGKRFQAQSALQQH H (part #1) (part #2) (part #3) 520 523 540 1/1 14/14 1/1 C CNRTFPSHTALKRH H 434 ZINC 1 463 ZINC 1 492 ZINC 1 520 ZINC 1
 (part #2) (part #3) 548 551 568 1/1 14/14") 548 ZINC 1 (part #1) (part #2) (part #3) 548 551 568 1/1 14/14 1/1 C CGSCFRDESTLKSH H (part #1) (part #2) (part #3) 576 579 596 1/1 14/14 1/1 C CDKKFSLKHQLETH H (part #1) (part #2) (part #3) 604 607 624 1/1 14/14 1/1 C CHQRSRDYSAMIKH H (part #1) (part #2) (part #3) 632 635 652 1/1 14/14 1/1 C CTEYCPSLSSMQKH H 576 ZINC 1 604 ZINC 1 632 ZINC 1 C 2 H 2 zinc finger motif
548 ZINC 1 (part #1) (part #2) (part #3) 548 551 568 1/1 14/14 1/1 C CGSCFRDESTLKSH H (part #1) (part #2) (part #3) 576 579 596 1/1 14/14 1/1 C CDKKFSLKHQLETH H (part #1) (part #2) (part #3) 604 607 624 1/1 14/14 1/1 C CHQRSRDYSAMIKH H (part #1) (part #2) (part #3) 632 635 652 1/1 14/14 1/1 C CTEYCPSLSSMQKH H 576 ZINC 1 604 ZINC 1 632 ZINC 1 C 2 H 2 zinc finger motif
 BLOCK ID AC DT DE PA ID ZINC_FINGER_C 2 H 2; BLOCK AC BL 00028; distance from previous block=(7, 2235) DE Zinc finger, C 2 H 2 type, domain proteins. BL CHP motif; width=29; seqs=135; 99. 5%=1594; strength=1246 ADR 1_YEAST ( 106) CEVCTRAFARQEHLKRHYRSHTNEKPYPC 10 AEF 1_DROME ( 214) CNVCDKTFRQSSTLTNHLKIHTGEKPYNC 10 AZF 1_YEAST ( 623) CDYCGKRFTQGGNLRTHERLHTGEKPYSC 10 BASO_HUMAN ( 358) CTACEKTFYDKGTLKIHYNAVHLKIKHKC 39 BRC 1_DROME ( 669) CNICKRVYSSLNSLRNHKSIYHRNLKQPK 37 BRC 2_DROME ( 471) CAICERVYCSRNSLMTHIYTYHKSRPGEM 27 BRC 3_DROME ( 467) GSLAAAVYSLHSHAHGHVLGHATSPPRPG 87 BRLA_EMENI ( 324) EPGCNGRFKRQEHLKRHMKSHSKEKPHVC 22 BTEB_RAT ( 147) YSGCGKVYGKSSHLKAHYRVHTGERPFPC 11 CF 23_DROME ( 368) CPDCPKTFKTPGTLAMHRKIHTGEAEREA 24 CF 2_DROME ( 403) CSYCGKSFTQSNTLKQHTRIHTGEKPFRC 11 ZINC_FINGER_C 2 H 2; PATTERN. Prosite PS 00028; APR-1990 (CREATED); JUN-1994 (DATA UPDATE); NOV-1995 (INFO UPDATE). Zinc finger, C 2 H 2 type, domain. C-x(2, 4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3, 5)-H.
BLOCK ID AC DT DE PA ID ZINC_FINGER_C 2 H 2; BLOCK AC BL 00028; distance from previous block=(7, 2235) DE Zinc finger, C 2 H 2 type, domain proteins. BL CHP motif; width=29; seqs=135; 99. 5%=1594; strength=1246 ADR 1_YEAST ( 106) CEVCTRAFARQEHLKRHYRSHTNEKPYPC 10 AEF 1_DROME ( 214) CNVCDKTFRQSSTLTNHLKIHTGEKPYNC 10 AZF 1_YEAST ( 623) CDYCGKRFTQGGNLRTHERLHTGEKPYSC 10 BASO_HUMAN ( 358) CTACEKTFYDKGTLKIHYNAVHLKIKHKC 39 BRC 1_DROME ( 669) CNICKRVYSSLNSLRNHKSIYHRNLKQPK 37 BRC 2_DROME ( 471) CAICERVYCSRNSLMTHIYTYHKSRPGEM 27 BRC 3_DROME ( 467) GSLAAAVYSLHSHAHGHVLGHATSPPRPG 87 BRLA_EMENI ( 324) EPGCNGRFKRQEHLKRHMKSHSKEKPHVC 22 BTEB_RAT ( 147) YSGCGKVYGKSSHLKAHYRVHTGERPFPC 11 CF 23_DROME ( 368) CPDCPKTFKTPGTLAMHRKIHTGEAEREA 24 CF 2_DROME ( 403) CSYCGKSFTQSNTLKQHTRIHTGEKPFRC 11 ZINC_FINGER_C 2 H 2; PATTERN. Prosite PS 00028; APR-1990 (CREATED); JUN-1994 (DATA UPDATE); NOV-1995 (INFO UPDATE). Zinc finger, C 2 H 2 type, domain. C-x(2, 4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3, 5)-H.
 Phylogenetic Analysis: Phylogenetics is the study of evolutionary relationships. Phylogenetic analysis is the means of inferring or estimating these relationship. The evolutionary history inferred from phylogenetic analysis is usually depicted as branching (treelike) diagrams, which represent a ort of pedigree of the inherited relationships among molecules (“gene trees”), organisms, or both. The four steps in phylogenetic analysis of DNA sequences are alignment, determining the substitution model, tree building, and tree evaluation. While other scientific analysis generally have empirical bases, phylogenetic analysis do not. The physical events yielding a phylogeny happened in the past, and can only be inferred or estimated. The three major tree-building criteria are distance, maximum parsimony, and maximum likelihood.
Phylogenetic Analysis: Phylogenetics is the study of evolutionary relationships. Phylogenetic analysis is the means of inferring or estimating these relationship. The evolutionary history inferred from phylogenetic analysis is usually depicted as branching (treelike) diagrams, which represent a ort of pedigree of the inherited relationships among molecules (“gene trees”), organisms, or both. The four steps in phylogenetic analysis of DNA sequences are alignment, determining the substitution model, tree building, and tree evaluation. While other scientific analysis generally have empirical bases, phylogenetic analysis do not. The physical events yielding a phylogeny happened in the past, and can only be inferred or estimated. The three major tree-building criteria are distance, maximum parsimony, and maximum likelihood.
 Over 130 packages available for various platforms
Over 130 packages available for various platforms
 Radial
Radial
 Slanted Cladogram
Slanted Cladogram
 Phylogram
Phylogram
 http: //www 2. ebi. ac. uk/clustalw/
http: //www 2. ebi. ac. uk/clustalw/
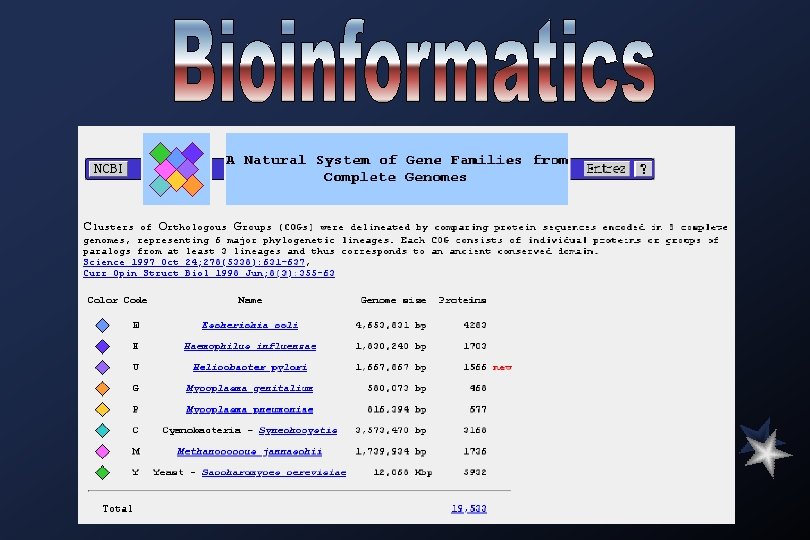
 Ortholog: Homologous genes that have diverged from each other after speciation events (e. g. , human beta- and chimp beta-globin) Paralog: Homologous genes that have diverged from each other after gene duplication events (e. g. , human beta- and gamma-globin) Xenolog: Homologous genes that have diverged from each other after lateral gene transfer events (e. g. , antibiotic resistance genes in bacteria) Homolog: Genes that are descended from a common ancestor (e. g. , all globins)
Ortholog: Homologous genes that have diverged from each other after speciation events (e. g. , human beta- and chimp beta-globin) Paralog: Homologous genes that have diverged from each other after gene duplication events (e. g. , human beta- and gamma-globin) Xenolog: Homologous genes that have diverged from each other after lateral gene transfer events (e. g. , antibiotic resistance genes in bacteria) Homolog: Genes that are descended from a common ancestor (e. g. , all globins)
 COG 0568 K DNA-dependent RNA polymerase sigma 70/sigma 32 subunits
COG 0568 K DNA-dependent RNA polymerase sigma 70/sigma 32 subunits
 EST: Expressed Sequences Tags db. EST is a division of Gen. Bank that contains sequence data and other information on "singlepass" c. DNA sequences, or Expressed Sequence Tags, from a number of organisms. There are 1, 775, 721 entries in human EST and 918, 414 entries in mouse EST. Total of 3, 643, 273 sequence entries in db. EST. (Feb. 18, 2000).
EST: Expressed Sequences Tags db. EST is a division of Gen. Bank that contains sequence data and other information on "singlepass" c. DNA sequences, or Expressed Sequence Tags, from a number of organisms. There are 1, 775, 721 entries in human EST and 918, 414 entries in mouse EST. Total of 3, 643, 273 sequence entries in db. EST. (Feb. 18, 2000).
 EST projects have their roots in the early 1980 s, when it was recognized that short stretches of DNA sequences from c. DNAs could be used to identify genes. The Institute for Genomic Research (TIGR) was established to generate EST data on a massive scale. Among the largest projects conducted entirely in the public domain include an effort funded by Merck and Co. , which has deposited more than 500, 000 human ESTs into db. EST. A hallmark of these endeavours, carried out by a collaboration between Washington University Genome Sequencing Center and members of IMAGE (Integrated Molecular Analysis of Gene Expression) consortium, has been the rapid deposition of the sequences into the public domain and the concomitant availability of the sequence-tagged clones.
EST projects have their roots in the early 1980 s, when it was recognized that short stretches of DNA sequences from c. DNAs could be used to identify genes. The Institute for Genomic Research (TIGR) was established to generate EST data on a massive scale. Among the largest projects conducted entirely in the public domain include an effort funded by Merck and Co. , which has deposited more than 500, 000 human ESTs into db. EST. A hallmark of these endeavours, carried out by a collaboration between Washington University Genome Sequencing Center and members of IMAGE (Integrated Molecular Analysis of Gene Expression) consortium, has been the rapid deposition of the sequences into the public domain and the concomitant availability of the sequence-tagged clones.
 db. EST release 021800 Summary by Organism - February 18, 2000 Number of public entries: 3, 643, 273 Homo sapiens (human) 1, 775, 721 Mus musculus + domesticus (mouse) 918, 414 Rattus sp. (rat) 134, 685 Caenorhabditis elegans (nematode) 101, 232 Drosophila melanogaster (fruit fly) 86, 121 Danio rerio (zebrafish) 61, 893 Lycopersicon esculentum (tomato) 53, 603 Zea mays (maize) 51, 883 Glycine max (soybean) 50, 656 Oryza sativa (rice) 47, 939 Arabidopsis thaliana (thale cress) 45, 757
db. EST release 021800 Summary by Organism - February 18, 2000 Number of public entries: 3, 643, 273 Homo sapiens (human) 1, 775, 721 Mus musculus + domesticus (mouse) 918, 414 Rattus sp. (rat) 134, 685 Caenorhabditis elegans (nematode) 101, 232 Drosophila melanogaster (fruit fly) 86, 121 Danio rerio (zebrafish) 61, 893 Lycopersicon esculentum (tomato) 53, 603 Zea mays (maize) 51, 883 Glycine max (soybean) 50, 656 Oryza sativa (rice) 47, 939 Arabidopsis thaliana (thale cress) 45, 757
 Search: AA 927876
Search: AA 927876
 db. EST Id: 1659486 IDENTIFIERS EST name: Gen. Bank Acc: Gen. Bank gi: om 18 b 09. s 1 AA 927876 3076620 CLONE INFO Clone Id: Source: Insert length: DNA type: IMAGE: 1541369 (3') NCI 1074 c. DNA PRIMERS Sequencing: SEQUENCE Quality: Entry Created: Last Updated: -40 m 13 fwd. ET from Amersham TTTGACGGGAGGGCACAGGAAACTCTTTATTATGGTGATGAGATCGACAATCTCCCCTAC TGTTAACCTTCGCTCCTGCACACTTCAGTGTCCTCACTCTGTAGGGCTCGCTGGCCTGGG CTTCTGCGACCCGCGATCGTCCAGGAGAGGGCACTCGGCGCCCTTCCTGGGGTNNTCTGG GGCGGAATTTGCTAGGCCGCCGTAGCAGCTGTGCCAGGTCAGAAGCCGGNCCGCT TTTCGTTCTTTAATTGGACTCTTGGCTAAGACGCTACCGACACCCCGTCAGTGGTGGAGG AAGAAGGACAACAGGGAGAGGTCGAGG High quality sequence stops at base: 318 Apr 17 1998 Jun 10 1998
db. EST Id: 1659486 IDENTIFIERS EST name: Gen. Bank Acc: Gen. Bank gi: om 18 b 09. s 1 AA 927876 3076620 CLONE INFO Clone Id: Source: Insert length: DNA type: IMAGE: 1541369 (3') NCI 1074 c. DNA PRIMERS Sequencing: SEQUENCE Quality: Entry Created: Last Updated: -40 m 13 fwd. ET from Amersham TTTGACGGGAGGGCACAGGAAACTCTTTATTATGGTGATGAGATCGACAATCTCCCCTAC TGTTAACCTTCGCTCCTGCACACTTCAGTGTCCTCACTCTGTAGGGCTCGCTGGCCTGGG CTTCTGCGACCCGCGATCGTCCAGGAGAGGGCACTCGGCGCCCTTCCTGGGGTNNTCTGG GGCGGAATTTGCTAGGCCGCCGTAGCAGCTGTGCCAGGTCAGAAGCCGGNCCGCT TTTCGTTCTTTAATTGGACTCTTGGCTAAGACGCTACCGACACCCCGTCAGTGGTGGAGG AAGAAGGACAACAGGGAGAGGTCGAGG High quality sequence stops at base: 318 Apr 17 1998 Jun 10 1998
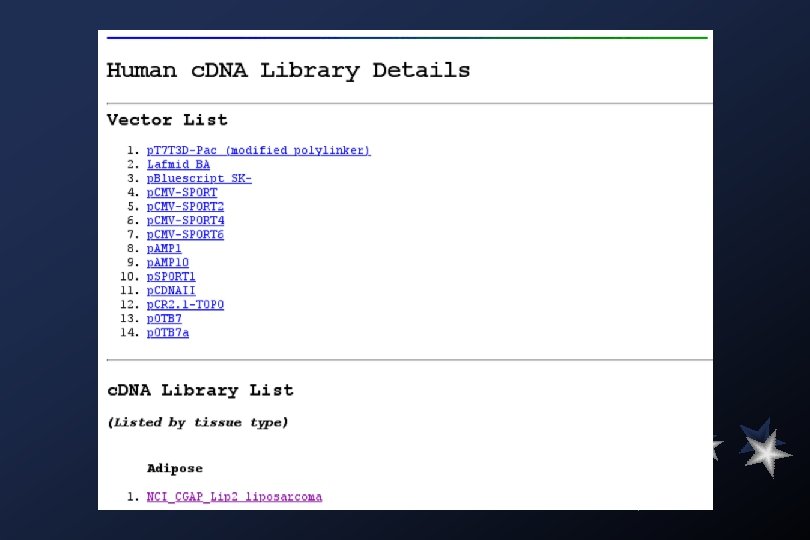
 COMMENTS This clone is available royalty-free through LLNL ; contact the IMAGE Consortium (info@image. llnl. gov) for further information. LIBRARY db. EST lib id: Lib Name: Organism: Organ: Lab host: Vector: R. Site 1: R. Site 2: Description: 1042 Soares_NFL_T_GBC_S 1 Homo sapiens pooled DH 10 B p. T 7 T 3 D-Pac (Pharmacia) with a modified polylinker Not I Eco RI Equal amounts of plasmid DNA from three normalized libraries (fetal lung Nb. HL 19 W, testis NHT, and B-cell NCI_CGAP_GCB 1) were mixed, and ss circles were made in vitro. Following HAP purification, this DNA was used as tracer in a subtractive hybridization reaction. The driver was PCR-amplified c. DNAs from pools of 5, 000 clones made from the same 3 libraries. The pools consisted of I. M. A. G. E. clones 297480 -302087, 682632 -687239, 726408 -728711, and 729096 -731399. Subtraction by Bento Soares and M. Fatima Bonaldo.
COMMENTS This clone is available royalty-free through LLNL ; contact the IMAGE Consortium (info@image. llnl. gov) for further information. LIBRARY db. EST lib id: Lib Name: Organism: Organ: Lab host: Vector: R. Site 1: R. Site 2: Description: 1042 Soares_NFL_T_GBC_S 1 Homo sapiens pooled DH 10 B p. T 7 T 3 D-Pac (Pharmacia) with a modified polylinker Not I Eco RI Equal amounts of plasmid DNA from three normalized libraries (fetal lung Nb. HL 19 W, testis NHT, and B-cell NCI_CGAP_GCB 1) were mixed, and ss circles were made in vitro. Following HAP purification, this DNA was used as tracer in a subtractive hybridization reaction. The driver was PCR-amplified c. DNAs from pools of 5, 000 clones made from the same 3 libraries. The pools consisted of I. M. A. G. E. clones 297480 -302087, 682632 -687239, 726408 -728711, and 729096 -731399. Subtraction by Bento Soares and M. Fatima Bonaldo.



 1, 775,") Simple Mathematics: Summary by Organism - February 18, 2000 Homo sapiens (human) 1, 775, 721 Human genes 100, 000 genes More than 10 fold coverage!!
Simple Mathematics: Summary by Organism - February 18, 2000 Homo sapiens (human) 1, 775, 721 Human genes 100, 000 genes More than 10 fold coverage!!
 Clustering is the process of finding subsets of sequences which belong together within a larger set. This is done by converting discrete similarity scores to boolean links between sequences. That is, two sequences are considered linked if their similarity exceeds a threshold. Uni. Gene clustering proceeds in several stages, with each stage adding less reliable data to the results of the preceding stage. This staged clustering affords greater control than a more egalitarian treatment of all links between sequences. Unigene_HUMAN: 92, 571 clusters| HGI: 299, 412 clusters Unigene_MOUSE: 75, 963 clusters| MGI: 104, 927 clusters Unigene_RAT: 28, 680 clusters | RGI: 35, 875 clusters (Feb. 19 , 2000) (Jul. 3, 1999)
Clustering is the process of finding subsets of sequences which belong together within a larger set. This is done by converting discrete similarity scores to boolean links between sequences. That is, two sequences are considered linked if their similarity exceeds a threshold. Uni. Gene clustering proceeds in several stages, with each stage adding less reliable data to the results of the preceding stage. This staged clustering affords greater control than a more egalitarian treatment of all links between sequences. Unigene_HUMAN: 92, 571 clusters| HGI: 299, 412 clusters Unigene_MOUSE: 75, 963 clusters| MGI: 104, 927 clusters Unigene_RAT: 28, 680 clusters | RGI: 35, 875 clusters (Feb. 19 , 2000) (Jul. 3, 1999)
 THCs, "Tentative Human Consensus" sequences, are assemblies of human ESTs. TIGR's Human Gene Index compare with Uni. Gene? The HGI assemblies (and all of TIGR's Gene Index assemblies) are made by first clustering the EST sequences and then assembling these clusters into consensus sequences, or THCs(TCs for non-human data). EST sequences are compared and clustered together if they meet the following criteria: a minimum 40 base pair match greater than 95% similarity in the overlap region a maximum unmatched overhang of 20 base pairs These clusters are then assembled into consensus sequences using TIGR's in-house assembly program. Uni. Gene links ESTs in a cluster if the sequences have a 50 base pair overlap in the 3' untranslated region (UTR) with 100% identity. These clusters are not run through the more stringent assembly process and consensus sequences are not made. For this reason you will often find several TIGR THCs contained within one Uni. Gene cluster.
THCs, "Tentative Human Consensus" sequences, are assemblies of human ESTs. TIGR's Human Gene Index compare with Uni. Gene? The HGI assemblies (and all of TIGR's Gene Index assemblies) are made by first clustering the EST sequences and then assembling these clusters into consensus sequences, or THCs(TCs for non-human data). EST sequences are compared and clustered together if they meet the following criteria: a minimum 40 base pair match greater than 95% similarity in the overlap region a maximum unmatched overhang of 20 base pairs These clusters are then assembled into consensus sequences using TIGR's in-house assembly program. Uni. Gene links ESTs in a cluster if the sequences have a 50 base pair overlap in the 3' untranslated region (UTR) with 100% identity. These clusters are not run through the more stringent assembly process and consensus sequences are not made. For this reason you will often find several TIGR THCs contained within one Uni. Gene cluster.
 Uni. Gene Human Release Statistics for Uni. Gene build uploaded on: Sat Feb 19 2000 Uni. Gene Build #108 Sequences Included in Uni. Gene =============== Known genes are from Gen. Bank 114 (1 -Dec-1999) ESTs are from db. EST through 13 -Feb-2000 30044 m. RNAs + gene CDSs 938584 EST, 3'reads 347845 EST, 5'reads + 157255 EST, other/unknown -----1473728 total sequences in clusters Final Number of Clusters (sets) ================ 92571 sets total 10797 sets contain at least one known gene 91523 sets contain at least one EST 9749 sets contain both genes and ESTs
Uni. Gene Human Release Statistics for Uni. Gene build uploaded on: Sat Feb 19 2000 Uni. Gene Build #108 Sequences Included in Uni. Gene =============== Known genes are from Gen. Bank 114 (1 -Dec-1999) ESTs are from db. EST through 13 -Feb-2000 30044 m. RNAs + gene CDSs 938584 EST, 3'reads 347845 EST, 5'reads + 157255 EST, other/unknown -----1473728 total sequences in clusters Final Number of Clusters (sets) ================ 92571 sets total 10797 sets contain at least one known gene 91523 sets contain at least one EST 9749 sets contain both genes and ESTs
 HGI Release 4. 5 - Nov. 15, 1999 Total sequences in THCs ESTs 1, 066, 183 HTs 5, 949 Totals 1, 072, 132 singletons 241, 110 1, 165 242, 275 Total unique sequences THCs singleton ESTs singleton HTs Total 84, 837 241, 110 1, 165 327, 112 total 1, 307, 293 7, 114 1, 314, 407
HGI Release 4. 5 - Nov. 15, 1999 Total sequences in THCs ESTs 1, 066, 183 HTs 5, 949 Totals 1, 072, 132 singletons 241, 110 1, 165 242, 275 Total unique sequences THCs singleton ESTs singleton HTs Total 84, 837 241, 110 1, 165 327, 112 total 1, 307, 293 7, 114 1, 314, 407

 Database: Unigene_HUMAN 58, 791 sequences; 43, 055, 747") AA 927876 as query (318 bps) Database: Unigene_HUMAN 58, 791 sequences; 43, 055, 747 total letters Score E Sequences producing significant alignments: (bits) Value gnl|UG|Hs#S 971963 ak 43 b 04. s 1 Homo sapiens c. DNA, 3' end /clone=IM. . . 599 e-171 gnl|UG|Hs#S 510257 70 F 12 Homo sapiens c. DNA /clone=(not-directiona. . . 36 0. 17 gnl|UG|Hs#S 971963 ak 43 b 04. s 1 Homo sapiens c. DNA, 3' end /clone=IMAGE: 1408687 /clone_end=3' /gb=AA 868505 /ug=Hs. 99430 /len=627 Length = 627 Score = 599 bits (302), Expect = e-171 Identities = 321/327 (98%), Positives = 321/327 (98%) Hs. 99430
AA 927876 as query (318 bps) Database: Unigene_HUMAN 58, 791 sequences; 43, 055, 747 total letters Score E Sequences producing significant alignments: (bits) Value gnl|UG|Hs#S 971963 ak 43 b 04. s 1 Homo sapiens c. DNA, 3' end /clone=IM. . . 599 e-171 gnl|UG|Hs#S 510257 70 F 12 Homo sapiens c. DNA /clone=(not-directiona. . . 36 0. 17 gnl|UG|Hs#S 971963 ak 43 b 04. s 1 Homo sapiens c. DNA, 3' end /clone=IMAGE: 1408687 /clone_end=3' /gb=AA 868505 /ug=Hs. 99430 /len=627 Length = 627 Score = 599 bits (302), Expect = e-171 Identities = 321/327 (98%), Positives = 321/327 (98%) Hs. 99430
 Hs. 99430 Homo sapiens EXPRESSION INFORMATION c. DNA sources: Blood, Ovary, Testis EST SEQUENCES (8) AI 150041 c. DNA clone IMAGE: 1751830 AA 927876 c. DNA clone IMAGE: 1541369 AI 223414 c. DNA clone IMAGE: 1838461 AI 150330 c. DNA clone IMAGE: 1751988 AA 868505 c. DNA clone IMAGE: 1408687 AA 476210 c. DNA clone IMAGE: 771312 AA 456628 c. DNA clone IMAGE: 809583 AI 361709 c. DNA clone IMAGE: 2021901 Testis 3' 3' Testis 3' Ovary 3' Blood 3' read 1. 1 read 1. 0 read 0. 6 read kb kb
Hs. 99430 Homo sapiens EXPRESSION INFORMATION c. DNA sources: Blood, Ovary, Testis EST SEQUENCES (8) AI 150041 c. DNA clone IMAGE: 1751830 AA 927876 c. DNA clone IMAGE: 1541369 AI 223414 c. DNA clone IMAGE: 1838461 AI 150330 c. DNA clone IMAGE: 1751988 AA 868505 c. DNA clone IMAGE: 1408687 AA 476210 c. DNA clone IMAGE: 771312 AA 456628 c. DNA clone IMAGE: 809583 AI 361709 c. DNA clone IMAGE: 2021901 Testis 3' 3' Testis 3' Ovary 3' Blood 3' read 1. 1 read 1. 0 read 0. 6 read kb kb
 Hs. 434 Homo sapiens Human heregulin-beta 1 gene, complete cds MAPPING INFORMATION Chromosome: 8 Gene Map 98: st. SG 4083 , Chr. 8, D 8 S 1820 -D 8 S 505 Gene Map 98: WI-18803 , Chr. 8, D 8 S 1820 -D 8 S 505 Gene Map 98: SHGC-12780 , Chr. 8, D 8 S 1820 -D 8 S 505 EXPRESSION INFORMATION c. DNA sources: Brain, Breast, Liver, Testis
Hs. 434 Homo sapiens Human heregulin-beta 1 gene, complete cds MAPPING INFORMATION Chromosome: 8 Gene Map 98: st. SG 4083 , Chr. 8, D 8 S 1820 -D 8 S 505 Gene Map 98: WI-18803 , Chr. 8, D 8 S 1820 -D 8 S 505 Gene Map 98: SHGC-12780 , Chr. 8, D 8 S 1820 -D 8 S 505 EXPRESSION INFORMATION c. DNA sources: Brain, Breast, Liver, Testis
 Database: HGI-HUMAN 234, 459 sequences; 111, 134, 950") AA 927876 as query (318 bps) Database: HGI-HUMAN 234, 459 sequences; 111, 134, 950 total letters Sequences producing significant alignments: Score (bits) E Value 581 40 e-165 0. 027 lcl|THC 226049 lcl|R 47793 34 1. 7 lcl|THC 226049 Length = 436 THC 226049 Score = 581 bits (293), Expect = e-165 Identities = 313/320 (97%), Positives = 313/320 (97%), Gaps = 1/320 (0%)
AA 927876 as query (318 bps) Database: HGI-HUMAN 234, 459 sequences; 111, 134, 950 total letters Sequences producing significant alignments: Score (bits) E Value 581 40 e-165 0. 027 lcl|THC 226049 lcl|R 47793 34 1. 7 lcl|THC 226049 Length = 436 THC 226049 Score = 581 bits (293), Expect = e-165 Identities = 313/320 (97%), Positives = 313/320 (97%), Gaps = 1/320 (0%)
 >THC 226049 TGAGGGCACAGGAAACTCTTTATTATGGTGATGAGATCGACAATCTCCCCTACTGTTAACCTTCGCTCCTGCACACTTCA GTGTCCTCACTCTGTAGGGCTCGCTGGCCTGGGCTTCTGCGACCCGCGATCGTCCAGGAGAGGGCACTCGGCGCCCTTCC TGGGGCg. TTc. TGGGGCGGAATTTGCTAGGCCGCCGTAGCAGCGGTGCCAGGTCAGAAGCCGGCy. CGCTTTTCGTT CTTTAATTGGACTCTTGGCTAAGACGCTACCGACACCCCGTCa. Gg. TGGTGGAGGAAGAAGGACAACAGGGAGAGGTCGAG GGCCGAGACGGCTCGAGGAGTAGAGGAAGGTGGAGCGGATGGTCCATCCGGGAGTTGGCTGGGCGAGTGACC GCGCATGTGCCGCTGCATGGAGGGCAAGCTGTTACA 1=================THC 226049================436 --------------1 --------------> -------------------2 -------------------> # EST Id GB# ATCC# left right library ----------------------------------------1 F zw 35 g 01. s 1 AA 476210 1 317 ovary tumor Nb. HOT, Soares 2 F zx 75 d 08. s 1 AA 456628 1 436 ovary tumor Nb. HOT, Soares Sequence source codes: F = Wash. U/Merck There are no hits for THC 226049.
>THC 226049 TGAGGGCACAGGAAACTCTTTATTATGGTGATGAGATCGACAATCTCCCCTACTGTTAACCTTCGCTCCTGCACACTTCA GTGTCCTCACTCTGTAGGGCTCGCTGGCCTGGGCTTCTGCGACCCGCGATCGTCCAGGAGAGGGCACTCGGCGCCCTTCC TGGGGCg. TTc. TGGGGCGGAATTTGCTAGGCCGCCGTAGCAGCGGTGCCAGGTCAGAAGCCGGCy. CGCTTTTCGTT CTTTAATTGGACTCTTGGCTAAGACGCTACCGACACCCCGTCa. Gg. TGGTGGAGGAAGAAGGACAACAGGGAGAGGTCGAG GGCCGAGACGGCTCGAGGAGTAGAGGAAGGTGGAGCGGATGGTCCATCCGGGAGTTGGCTGGGCGAGTGACC GCGCATGTGCCGCTGCATGGAGGGCAAGCTGTTACA 1=================THC 226049================436 --------------1 --------------> -------------------2 -------------------> # EST Id GB# ATCC# left right library ----------------------------------------1 F zw 35 g 01. s 1 AA 476210 1 317 ovary tumor Nb. HOT, Soares 2 F zx 75 d 08. s 1 AA 456628 1 436 ovary tumor Nb. HOT, Soares Sequence source codes: F = Wash. U/Merck There are no hits for THC 226049.
 In silico cloning: In order to perform an electronic c. DNA library screen, the EST sequences retrieved in this way can be used as queries in a BLASTN search of db. EST to identify over-lapping ESTs. This procedure can be reiterated with the newly identified ESTs until no additional hits are found. The ESTs isolated can be assembled into sequence contigs using computer softwares. How to start? TBLASTN emb|AJ 003623|HSJ 003623 H. sapiens DNA for EST MPIpl 10 -4 B 1 Length = 556 Score = 46. 9 bits (109), Expect = 1 e-04 Identities = 29/83 (34%), Positives = 42/83 (49%), Gaps = 8/83 (9%) Query: 23 RLRPALCDTLITVGSQEFPAHSLVLAGVSQQLGRRGQWALGEG----ISPSTFAQL 74 RL+ LCD L+ VG Q+F AH VLA S+ E P F + Sbjct: 164 RLKGQLCDVLLIVGDQKFRAHKNVLAASSEYFQSLFTNKENESQTVFQLDFCEPDAFDNV 3 Query: 75 LNFVYGESVELQPGELRPLQEAARALGVQSL 105 LN++Y S+ ++ L +QE +LG+ L Sbjct: 344 LNYIYSSSLFVEKSSLAAVQELGYSLGISFL 436
In silico cloning: In order to perform an electronic c. DNA library screen, the EST sequences retrieved in this way can be used as queries in a BLASTN search of db. EST to identify over-lapping ESTs. This procedure can be reiterated with the newly identified ESTs until no additional hits are found. The ESTs isolated can be assembled into sequence contigs using computer softwares. How to start? TBLASTN emb|AJ 003623|HSJ 003623 H. sapiens DNA for EST MPIpl 10 -4 B 1 Length = 556 Score = 46. 9 bits (109), Expect = 1 e-04 Identities = 29/83 (34%), Positives = 42/83 (49%), Gaps = 8/83 (9%) Query: 23 RLRPALCDTLITVGSQEFPAHSLVLAGVSQQLGRRGQWALGEG----ISPSTFAQL 74 RL+ LCD L+ VG Q+F AH VLA S+ E P F + Sbjct: 164 RLKGQLCDVLLIVGDQKFRAHKNVLAASSEYFQSLFTNKENESQTVFQLDFCEPDAFDNV 3 Query: 75 LNFVYGESVELQPGELRPLQEAARALGVQSL 105 LN++Y S+ ++ L +QE +LG+ L Sbjct: 344 LNYIYSSSLFVEKSSLAAVQELGYSLGISFL 436
 Experimental results: TTGANNNCCTTTGAANNNCCNNTTNNTCATAGATCTCTCGAGTTTTTTTTTTCTGAAGGGCACAGGAAAC TCTTTATTATGGTGATGAGATCGACAATCTCCCCTACTGTTAACCTTCGCTCCTGCACACTTCAGTGTCCTCACTCTGTAGGG CTCGCTGGCCTGGGCTTCTGCGACCCGCGATCGTCCAGGAGAGGGCACTCGGCGCCCTTCCTGGGGCGCTTCTGGGGCGGAAT TTGCTAGGCCGCCGTAGCAGCGGTGCCAGGTCAGAAGCCGGCCCGCTTTTCGTTCTTTAATTGGACTCTTGGCTAAGA CGCTACCGACACCCCGTCAGGTGGTGGAGGAAGAAGGACAACAGGGAGAGGTCGAGGGCCGAGACGGCCTCGAGGAGGAGTAG AGGAAGGTGGAGCGGATGGTCCATCCGGGAGTTGGCTGGGCGAGTGACCGCGCATGTGCGCCTGCATGGAGGCCAGGCT GGGACAGCCGGCCCCGCACAGGGAGCAGCGGTACGGAGCGGCCCCGTGTGTCCGCAGGTGCTTGGTCATGGCCGAGAAGTCCC GGGAGCGCTGAGGACAAAGGCTACAGGAGAAGGGCTTCTCTCCTGTGTGGACTCGGTAGTGCGTCTCCATCTGATGCTTGAGT GAAAACCTCTTTCACAGAGCACGCATAGGGGCCCAGACCGAGCANGGTCGACGCGGCCCGCGAAATTCGGATCCCCGGG GCCTTCATGGGCCATATGACCCCCCAAGCTAGCGTAAATCTGGGAACATCGTATGGGTAAAGCCNTNANAGAATCTCTTTTTT TTTGGGGNGGGGGTNATCTTTCATTNATCGAATTAGANTAGTTATNTNCCATTAATCCATTGNANNGGNNTTTAAAC ATTCCCTTGAAGGGATTCCNAAACCCTTTTACCNCAATTTTGGGTCCCGTCCAAACCCAGGTTGACAAGNGGGTTTTTGGAAA TTNTTTNCCCNTNATTCAATTTTTCCT Yeast two-hybrid experiment; Differential Display; Library screening; etc.
Experimental results: TTGANNNCCTTTGAANNNCCNNTTNNTCATAGATCTCTCGAGTTTTTTTTTTCTGAAGGGCACAGGAAAC TCTTTATTATGGTGATGAGATCGACAATCTCCCCTACTGTTAACCTTCGCTCCTGCACACTTCAGTGTCCTCACTCTGTAGGG CTCGCTGGCCTGGGCTTCTGCGACCCGCGATCGTCCAGGAGAGGGCACTCGGCGCCCTTCCTGGGGCGCTTCTGGGGCGGAAT TTGCTAGGCCGCCGTAGCAGCGGTGCCAGGTCAGAAGCCGGCCCGCTTTTCGTTCTTTAATTGGACTCTTGGCTAAGA CGCTACCGACACCCCGTCAGGTGGTGGAGGAAGAAGGACAACAGGGAGAGGTCGAGGGCCGAGACGGCCTCGAGGAGGAGTAG AGGAAGGTGGAGCGGATGGTCCATCCGGGAGTTGGCTGGGCGAGTGACCGCGCATGTGCGCCTGCATGGAGGCCAGGCT GGGACAGCCGGCCCCGCACAGGGAGCAGCGGTACGGAGCGGCCCCGTGTGTCCGCAGGTGCTTGGTCATGGCCGAGAAGTCCC GGGAGCGCTGAGGACAAAGGCTACAGGAGAAGGGCTTCTCTCCTGTGTGGACTCGGTAGTGCGTCTCCATCTGATGCTTGAGT GAAAACCTCTTTCACAGAGCACGCATAGGGGCCCAGACCGAGCANGGTCGACGCGGCCCGCGAAATTCGGATCCCCGGG GCCTTCATGGGCCATATGACCCCCCAAGCTAGCGTAAATCTGGGAACATCGTATGGGTAAAGCCNTNANAGAATCTCTTTTTT TTTGGGGNGGGGGTNATCTTTCATTNATCGAATTAGANTAGTTATNTNCCATTAATCCATTGNANNGGNNTTTAAAC ATTCCCTTGAAGGGATTCCNAAACCCTTTTACCNCAATTTTGGGTCCCGTCCAAACCCAGGTTGACAAGNGGGTTTTTGGAAA TTNTTTNCCCNTNATTCAATTTTTCCT Yeast two-hybrid experiment; Differential Display; Library screening; etc.
 BLASTN search to Gen. Bank
BLASTN search to Gen. Bank
 Cosmid from chromosome 19; it is a novel gene.
Cosmid from chromosome 19; it is a novel gene.
 BLASTN search to db. EST; Unigene; TIGR-HGI
BLASTN search to db. EST; Unigene; TIGR-HGI
 c. DNA and genomic DNA alignment and matrix analysis:
c. DNA and genomic DNA alignment and matrix analysis:
 Gene prediction programs:
Gene prediction programs:
 http: //CCR-081. mit. edu/GENSCAN. html
http: //CCR-081. mit. edu/GENSCAN. html
 GRAIL 2 10138 - 11018 12608 - 12748 13530 - 13923 + x x GENSCAN 10138 - 11018 11268 - 11341 11450 - 11518 11644 - 11808 11989 - 12144 12360 - 12454 12608 - 12748 + + + x x FGENES 1880 - 1908 5061 - 5175 5900 - 6049 8317 - 8544 10357 - 11018 11268 - 11341 11450 - 11518 11644 - 11864 poly. A: 12521 x x x + + +
GRAIL 2 10138 - 11018 12608 - 12748 13530 - 13923 + x x GENSCAN 10138 - 11018 11268 - 11341 11450 - 11518 11644 - 11808 11989 - 12144 12360 - 12454 12608 - 12748 + + + x x FGENES 1880 - 1908 5061 - 5175 5900 - 6049 8317 - 8544 10357 - 11018 11268 - 11341 11450 - 11518 11644 - 11864 poly. A: 12521 x x x + + +
 ATGTCCCTGCCCCCCATAAGACTGCCCAGCCCCTATGGCTCTGATCGGCTGGTACAGCTAGCAGCCAGGCTCCGGCCAGCACTCTGTGATACTCTGATCACCGTAGGGAGCCAGGAGTTC M S L P P I R L P S P") (Start) ATGTCCCTGCCCCCCATAAGACTGCCCAGCCCCTATGGCTCTGATCGGCTGGTACAGCTAGCAGCCAGGCTCCGGCCAGCACTCTGTGATACTCTGATCACCGTAGGGAGCCAGGAGTTC M S L P P I R L P S P Y G S D R L V Q L A A R L R P A L C D T L I T V G S Q E F> CCCGCCCACAGCCTGGTGCTAGCAGGTGTCAGCAGCTGGGCCGCAGGGGCCAGTGGGCTCTGGGAGAAGGCATCAGCCCTTCTACCTTTGCCCAGCTCCTGAACTTTGTGTATGGG P A H S L V L A G V S Q Q L G R R G Q W A L G E G I S P S T F A Q L L N F V Y G> GAGAGTGTAGAGCTGCAGCCTGGAGAGCTAAGGCCCCTTCAGGAGGCGGCCAGGGCCTTGGGAGTGCAGTCCCTGGAAGAGGCATGCTGGAGGGCTCGAGGGGACAGGGCTAAAAAGCCA E S V E L Q P G E L R P L Q E A A R A L G V Q S L E E A C W R A R G D R A K K P> GATCCAGGCCTGAAGAAACATCAGGAGGAGCCAGAGAAACCCTCAAGGAATCCTGAGAACTGGGGGACCCTGGAGAGAAGCAGAAACCAGAACAGGTTTCTAGAACTGGTGGGAGA D P G L K K H Q E E P E K P S R N P E R E L G D P G E K Q K P E Q V S R T G G R> GAACAGGAGATGTTGCACAAGCACTCGCCACCAAGAGGCAGACCCGAGATGGCAGGAGCAACGCAGGAGGCTCAGCAGGAACAGACCAGGTCAAAGGAGAAACGCCTCCAAGCCCCTGTT E Q E M L H K H S P P R G R P E M A G A T Q E A Q Q E Q T R S K E K R L Q A P V> GGCCAAAGGGGAGCAGATGGGAAGCATGGAGTGCTCACGTGGTTGAGGGAAAATCCAGGGGGCTCTGAGGAAAGTCTGCGCAAGCTCCCTGGCCCCCTTCCCCCAGCAGGCTCCCTGCAA G Q R G A D G K H G V L T W L R E N P G G S E E S L R K L P G P L P P A G S L Q> ACCAGCGTCACCCCTAGGCCCTCGTGGGCTGAGGCCCCTTGGTGGGGGGCCAGCCTGCCCTGTGGAGCATCCTGCTGATGCCGCCCAGATATGGCATTCCCTTCTACCATAGCACC T S V T P R P S W A E A P W L V G G Q P A L W S I L L M P P R Y G I P F Y H S T> CCCACCACTGGAGCCTGGCAGGAGGTCTGGCGGGAACAGAGGATCCCACTGTCCCTAAATGCCCCCAAAGGGCTCTGGAGCCAGAACCAGTTGGCCTCCTCCAGCCCTACCCCAGGTTCC P T T G A W Q E V W R E Q R I P L S L N A P K G L W S Q N Q L A S S S P T P G S> CTCCCCCAGGGCCCCGCACAGCTCAGCCCTGGGGAGATGGAAGAGTCTGATCAGGGGCACACAGGCGCACTTGCAACCTGTGCGGGTCATGAGGACAAGGCTGCCCACCTCGCCCG L P Q G P A Q L S P G E M E E S D Q G H T G A L A T C A G H E D K A G C P P R P> CACCCTCCCCCGGCCCCTCCTGCTCGGTCTCGGCCCTATGCGTGCTCTGTGGAAAGAGGTTTTCACTCAAGCATCAGATGGAGACGCACTACCGAGTCCACACAGGAGAGAAGCCC H P P P A R S R P Y A C S V C G K R F S L K H Q M E T H Y R V H T G E K P> TTCTCCTGTAGCCTTTGTCCTCAGCGCTCCCGGGACTTCTCGGCCATGACCAAGCACCTGCGGACACACGGGGCCGCTCCGTACCGCTGCTCCCTGTGCGGGGCCGGCTGTCCCAGCCTG F S C S L C P Q R S R D F S A M T K H L R T H G A A P Y R C S L C G A G C P S L> GCCTCCATGCAGGCGCACATGCGCGGTCACTCGCCCAGCCAACTCCCGGATGGACCATCCGCTCCACCTTCCTCTACTCCTCCTCGAGGCCGTCTCGGCCCTCGACCTCTCCCTGT A S M Q A H M R G H S P S Q L P P G W T I R S T F L Y S S S R P S T S P C> TGTCCTTCTTCCTCCACCACCTGACGGGGTGTCGGTAGCGTCTTAGCCAAGAGTCCAATTAAAGAACGAAAAGCGGGCCGGCTTCTGACCTGGCACCGCTGCTACGGCGGCCTAG C P S S S T T *
(Start) ATGTCCCTGCCCCCCATAAGACTGCCCAGCCCCTATGGCTCTGATCGGCTGGTACAGCTAGCAGCCAGGCTCCGGCCAGCACTCTGTGATACTCTGATCACCGTAGGGAGCCAGGAGTTC M S L P P I R L P S P Y G S D R L V Q L A A R L R P A L C D T L I T V G S Q E F> CCCGCCCACAGCCTGGTGCTAGCAGGTGTCAGCAGCTGGGCCGCAGGGGCCAGTGGGCTCTGGGAGAAGGCATCAGCCCTTCTACCTTTGCCCAGCTCCTGAACTTTGTGTATGGG P A H S L V L A G V S Q Q L G R R G Q W A L G E G I S P S T F A Q L L N F V Y G> GAGAGTGTAGAGCTGCAGCCTGGAGAGCTAAGGCCCCTTCAGGAGGCGGCCAGGGCCTTGGGAGTGCAGTCCCTGGAAGAGGCATGCTGGAGGGCTCGAGGGGACAGGGCTAAAAAGCCA E S V E L Q P G E L R P L Q E A A R A L G V Q S L E E A C W R A R G D R A K K P> GATCCAGGCCTGAAGAAACATCAGGAGGAGCCAGAGAAACCCTCAAGGAATCCTGAGAACTGGGGGACCCTGGAGAGAAGCAGAAACCAGAACAGGTTTCTAGAACTGGTGGGAGA D P G L K K H Q E E P E K P S R N P E R E L G D P G E K Q K P E Q V S R T G G R> GAACAGGAGATGTTGCACAAGCACTCGCCACCAAGAGGCAGACCCGAGATGGCAGGAGCAACGCAGGAGGCTCAGCAGGAACAGACCAGGTCAAAGGAGAAACGCCTCCAAGCCCCTGTT E Q E M L H K H S P P R G R P E M A G A T Q E A Q Q E Q T R S K E K R L Q A P V> GGCCAAAGGGGAGCAGATGGGAAGCATGGAGTGCTCACGTGGTTGAGGGAAAATCCAGGGGGCTCTGAGGAAAGTCTGCGCAAGCTCCCTGGCCCCCTTCCCCCAGCAGGCTCCCTGCAA G Q R G A D G K H G V L T W L R E N P G G S E E S L R K L P G P L P P A G S L Q> ACCAGCGTCACCCCTAGGCCCTCGTGGGCTGAGGCCCCTTGGTGGGGGGCCAGCCTGCCCTGTGGAGCATCCTGCTGATGCCGCCCAGATATGGCATTCCCTTCTACCATAGCACC T S V T P R P S W A E A P W L V G G Q P A L W S I L L M P P R Y G I P F Y H S T> CCCACCACTGGAGCCTGGCAGGAGGTCTGGCGGGAACAGAGGATCCCACTGTCCCTAAATGCCCCCAAAGGGCTCTGGAGCCAGAACCAGTTGGCCTCCTCCAGCCCTACCCCAGGTTCC P T T G A W Q E V W R E Q R I P L S L N A P K G L W S Q N Q L A S S S P T P G S> CTCCCCCAGGGCCCCGCACAGCTCAGCCCTGGGGAGATGGAAGAGTCTGATCAGGGGCACACAGGCGCACTTGCAACCTGTGCGGGTCATGAGGACAAGGCTGCCCACCTCGCCCG L P Q G P A Q L S P G E M E E S D Q G H T G A L A T C A G H E D K A G C P P R P> CACCCTCCCCCGGCCCCTCCTGCTCGGTCTCGGCCCTATGCGTGCTCTGTGGAAAGAGGTTTTCACTCAAGCATCAGATGGAGACGCACTACCGAGTCCACACAGGAGAGAAGCCC H P P P A R S R P Y A C S V C G K R F S L K H Q M E T H Y R V H T G E K P> TTCTCCTGTAGCCTTTGTCCTCAGCGCTCCCGGGACTTCTCGGCCATGACCAAGCACCTGCGGACACACGGGGCCGCTCCGTACCGCTGCTCCCTGTGCGGGGCCGGCTGTCCCAGCCTG F S C S L C P Q R S R D F S A M T K H L R T H G A A P Y R C S L C G A G C P S L> GCCTCCATGCAGGCGCACATGCGCGGTCACTCGCCCAGCCAACTCCCGGATGGACCATCCGCTCCACCTTCCTCTACTCCTCCTCGAGGCCGTCTCGGCCCTCGACCTCTCCCTGT A S M Q A H M R G H S P S Q L P P G W T I R S T F L Y S S S R P S T S P C> TGTCCTTCTTCCTCCACCACCTGACGGGGTGTCGGTAGCGTCTTAGCCAAGAGTCCAATTAAAGAACGAAAAGCGGGCCGGCTTCTGACCTGGCACCGCTGCTACGGCGGCCTAG C P S S S T T *