Генные и хром. заболвания.ppt
- Количество слайдов: 47



ГЕННЫЕ И ХРОМОСОМНЫЕ ЗАБОЛЕВАНИЯ Кафедра детских болезней № 1 АО «МУА» Зав. каф. , д. м. н. , проф. Моренко М. А.

Определение l. Генные болезни – это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена

Эпидемиология Общая частота генных болезней в популяции составляет 1 -2%. Условно частоту генных болезней считают: 1. Высокой, если она встречается с частотой 1 случай на 10000 новорожденных; 2. Средней – 1 на 10000 – 40000; 3. Низкой (больше 40000).

Основная схема генных болезней включает ряд звеньев: l Мутантный аллель → измененный первичный l l l l продукт → цепь биохимических процессов в клетке → органы → организм В результате мутации гена на молекулярном уровне возможны следующие варианты: - синтез аномального белка; - выработка избыточного количества генного продукта; - отсутствие выработки первичного продукта; - выработка уменьшенного количества нормального первичного продукта.

Классификация l - Болезни аминокислотного обмена l - Нарушения обмена углеводов - Болезни, связанные с нарушением липидного обмена l - Наследственные болезни пуринового и l пиримидинового обмена l - Болезни нарушения обмена соединительной ткани l - Наследственные нарушения циркулирующих белков l - Болезни, связанные с нарушением обмена в l эритроцитах l - Наследственные болезни обмена металлов l - Синдромы нарушения всасывания в l пищеварительном тракте

Генные болезни l К генным болезням у человека относятся многочисленные болезни обмена веществ. l Они могут быть связаны с нарушением: l - обмена углеводов, l - липидов, l - стероидов, пуринов и пиримидинов, l - билирубина, l - металлов и др. l Пока еще нет единой классификации наследственных болезней обмена веществ.

Болезни обмена l Болезни аминокислотного обмена: l Фенилкетонурия l Нарушения обмена углеводов: l Галактоземия l Гликогеновая болезнь l Нарушение липидного обмена l Ниманна-Пика – снижение фермента сфирингомиелиназы (нарушение НС) l Болезнь Гоше - накопление цереброзидов в клетках нервной и РЭС, обусловленное дефицитом фермента глюкоцереброзидазы.
 l Синдром Марфана ( «паучьи пальцы» , арахнодактилия)")
Болезни нарушения обмена соединительной ткани (1) l Синдром Марфана ( «паучьи пальцы» , арахнодактилия) - поражение соединительной ткани вследствие мутации в гене, ответственном за синтез фибриллина; l Мукополисахаридозы - группа заболеваний соединительной ткани, связанных с нарушеним обмена кислых гликозаминогликанов.
 l. Фибродисплазия - заболевание соединительной ткани, связанное с")
Болезни нарушения обмена соединительной ткани (2) l. Фибродисплазия - заболевание соединительной ткани, связанное с ее прогрессирующим окостенением в результате мутации в гене ACVR 1

Наследственные болезни пуринового и пиримидинового обмена l Подагра; l Синдром Леша-Найхана. l Наследственные болезни обмена металлов l Болезнь Коновалова-Вильсона и др. l Синдромы нарушения всасывания в пищеварительном тракте l Муковисцидоз; l Непереносимость лактозы и др.
")
Нарушения циркулирующих белков l. Гемоглобинопатии - наследственные нарушения синтеза гемоглобина. l. Выделяют количественные (структурные) и качественные. l. Их формы остаются нормальной, снижена лишь скорость синтеза глобиновых цепей (талассемия).

Болезни, связанные с нарушением обмена в эритроцитах l. Гемолитические анемии - снижение уровня гемоглобина и укорочением срока жизни эритроцитов; l. Наследственный микросфероцитоз - врождённая недостаточность липидов оболочки эритроцитов.

Хромосомные болезни l Хромосомные болезни обусловлены грубым нарушением наследственного аппарата — изменением числа и структуры хромосом. l Типичная причина, в частности, — алкогольная интоксикация родителей при зачатии ( «пьяные дети» ). l К ним относятся: l Синдром Дауна, Клайнфельтера, Шерешевского — Тернера, Эдвардса «кошачьего крика» и др.

Общепопуляционная частота врожденных и наследственных нарушений l. Примерно 5 -6 детей из 100 новорожденных рождаются с какими-нибудь генетически обусловленными заболеваниями.

Диагностика l Диагностика производится следующими методами: l- цитогенетическим; l- молекулярно-цитогенетическими; l- молекулярно-генетическими; l- фенотипирование l (синдромологический подход) l

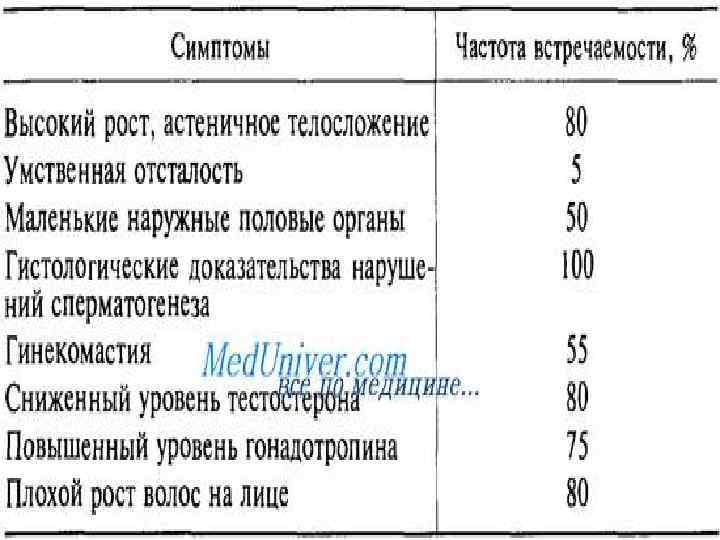
Синдром Клайнфелтера l Клиническая картина синдрома описана в 1942 году в работах Гарри Клайнфельтера и Фуллера Олбрайта. l Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их сочетаний (мозаицизм). l Обнаружено несколько типов полисомии по хромосомам X и Y у лиц мужского пола: 47, XXY; 47, XYY; 48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY; 49 XXXYY. l Наиболее распространен синдром Клайнфельтера (XXY). l Общая частота его колеблется в пределах 1 на 500700 новорожденных мальчиков.

Синдром Клайнфелтера l При синдроме Клайнфелтера независимо от числа дополнительных хромосом X определяет развитие мужского пола. l Самым частым является кариотип 47, XXY.
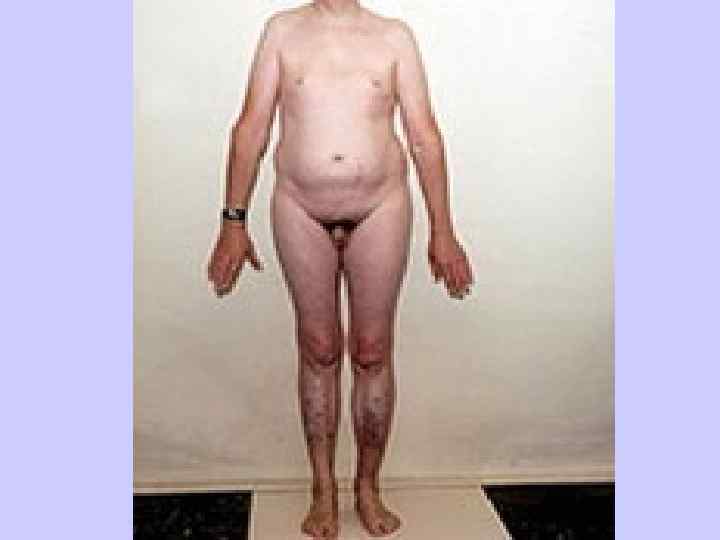

Синдром Шереше вского — Тернера Хромосомная болезнь, сопровождающаяся характерными: - - аномалиями физического - развития, - - низкорослостью; - - половым инфантилизмом. l

Синдром Шерешевского — Тернера Впервые эта болезнь как наследственная l была описана в 1925 г. Н. А. Шерешевским, который считал, что она обусловлена недоразвитием половых желез и передней доли гипофиза и сочетается с врожденными пороками внутреннего развития. l В 1938 г. Тернер выделил характерную для этого симптомокомплекса триаду симптомов: половой инфантилизм, кожные крыловидные складки на боковых поверхностях шеи и деформацию локтевых суставов.
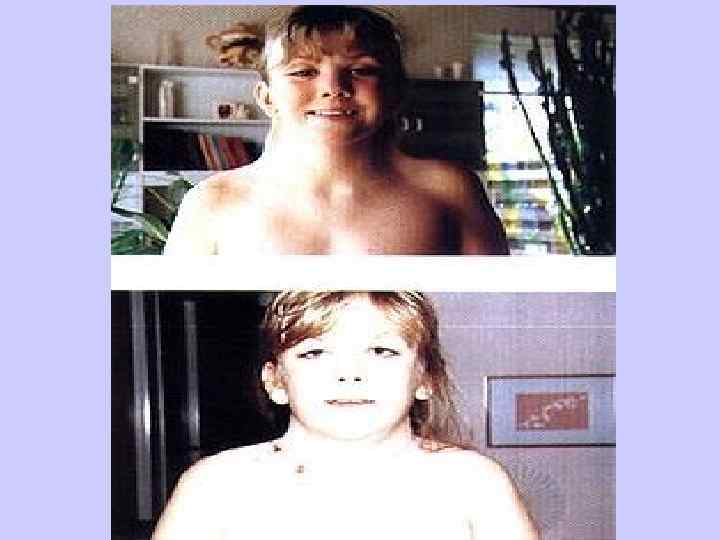

Синдром Патау l Трисомия 13, или синдром Патау, встречается с частотой 1 на 10 000 новорожденных. l Клинические проявления синдрома Патау, так же как и синдрома Эдвардса, как правило, очень тяжелые и включают множественные врожденные пороки развития.

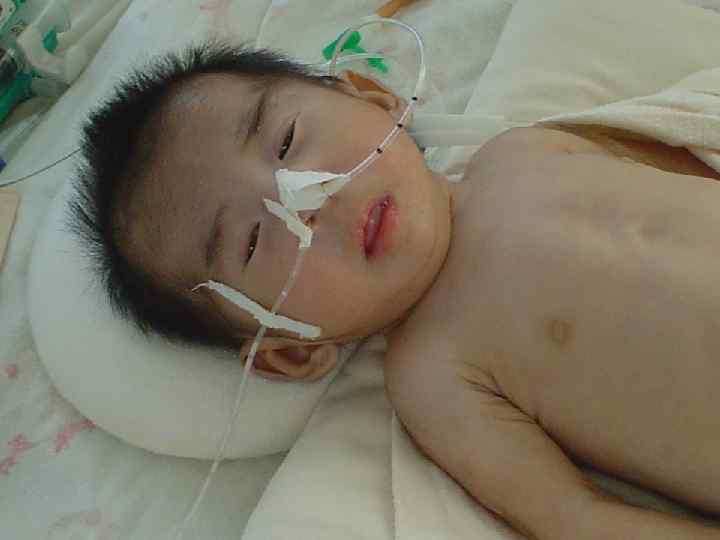
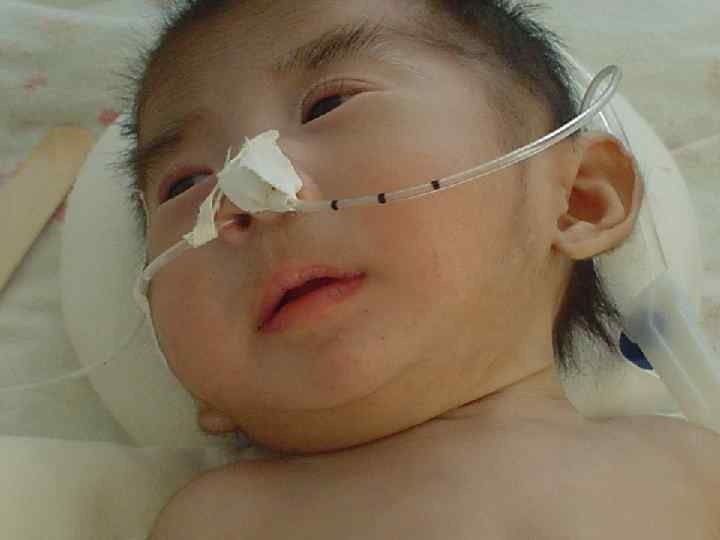
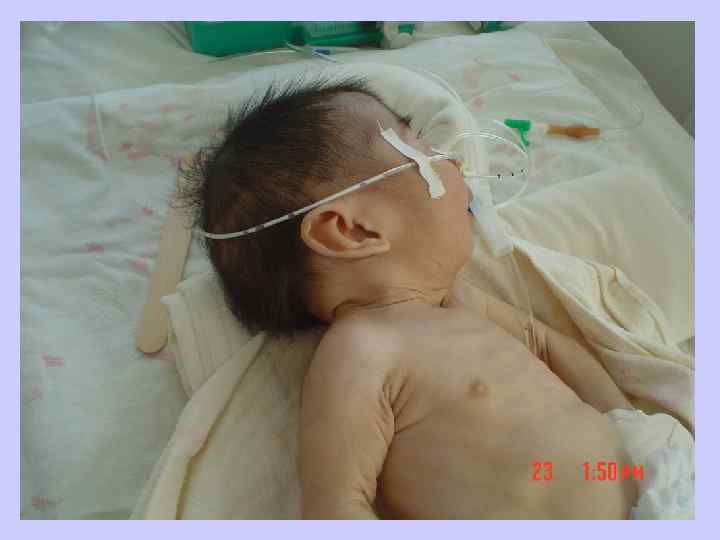
 — одна из")
Синдром Дауна l Синдро м Да уна (трисомия по хромосоме 21) — одна из форм геномной патологии, при которой чаще всего кариотип представлен 47 хромосомами вместо нормальных 46, поскольку хромосомы 21 -й пары, вместо нормальных двух, представлены тремя копиями (трисомия, также плоидность). l Существует ещё две формы данного синдрома: транслокация хромосомы 21 на другие хромосомы (чаще на 15, реже на 14, ещё реже на 21, 22 и Yхромосому) — 4 % случаев, и мозаичный вариант синдрома — 5 %.
,")
Синдром Дауна l Синдром получил название в честь английского врача Джона Дауна (John Down), впервые описавшего в 1866 году. l Связь между происхождением врождённого синдрома и изменением количества хромосом была выявлена только в 1959 году французским генетиком Жеромом Леженом. l Слово «синдром» означает набор признаков или характерных черт. При употреблении этого термина предпочтительнее форма «синдром Дауна» , а не «болезнь Дауна» . l Первый Международный день человека с синдромом Дауна был проведён 21 марта 2006 года.

Джон Лэнгдон Даун l В 1862 г. l В 1866 г. l В 1950 г.

Кариотип синдрома Дауна

Вероятность с-ма Дауна в зависимости от возраста матери
 — 81 % l Кожная складка")
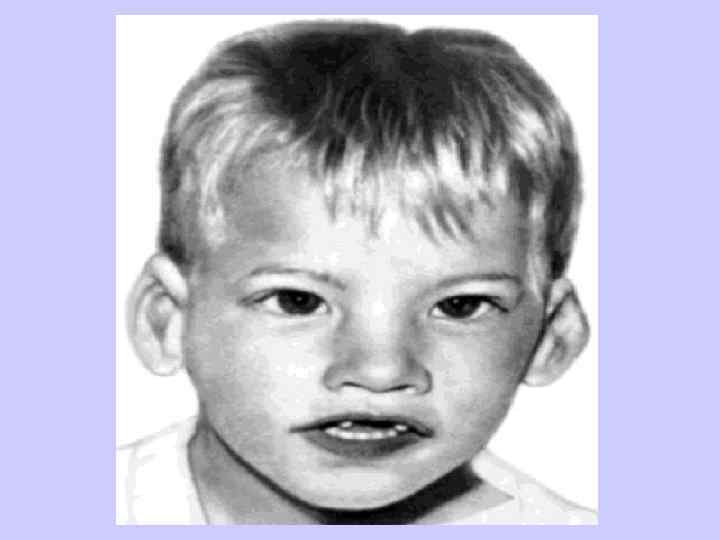
Клиника синдрома Дауна l Брахицефалия (аномальное укорочение черепа) — 81 % l Кожная складка на шее у новорожденных l — 81 % l Эпикантус (вертикальная кожная складка, прикрывающая медиальный угол глазной щели) — 80 % l Гиперподвижность суставов — 80 % l Мышечная гипотония — 80 % l Плоский затылок — 78 %

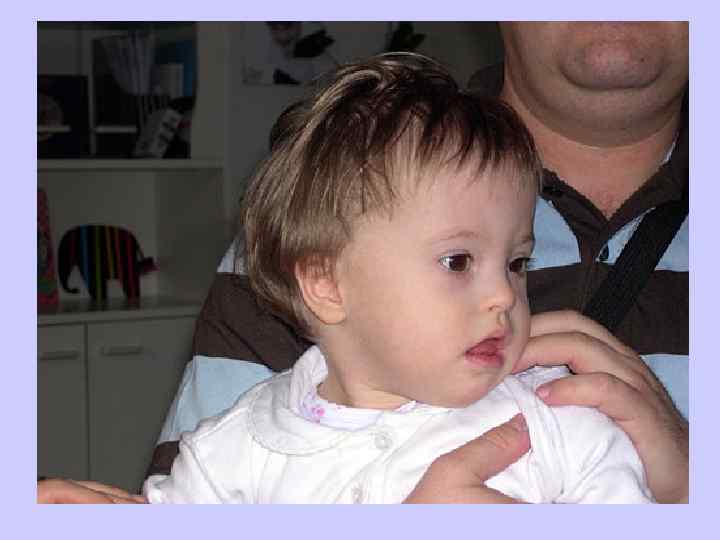
 — 45 %;")
Клиника болезни Дауна l Поперечная ладонная складка (называемая также «обезьяньей» ) — 45 %; l Короткая широкая шея — 45 %; l ВПС (врождённый порок сердца) — 40 %; l Короткий нос — 40 %; l Страбизм (косоглазие) — 29 %; l Деформация грудной клетки (килевидная или воронкообразная) — 27 %.
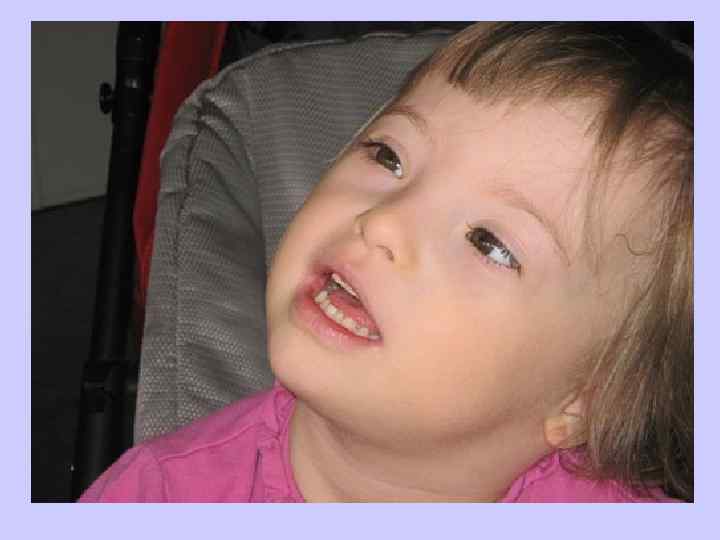

Клиника синдрома Дауна l - Короткие конечности - 70 %; l - Брахимезофалангия (укорочение всех l пальцев за счет недоразвития средних l фаланг) - 70 %; l - Катаракта в возрасте старше 8 лет - 66 %; l - Открытый рот (в связи с низким тонусом l мышц и особым строением нёба) - 65 %; l - Зубные аномалии — 65 %.
 — 60 %;")
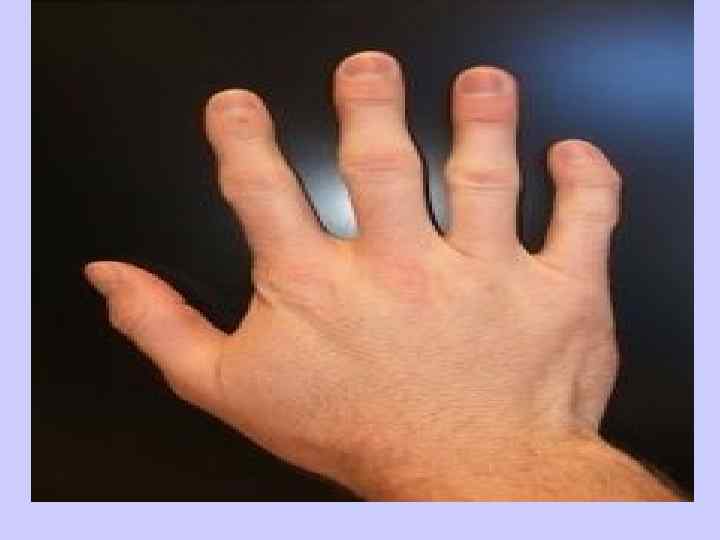
Клиника синдрома Дауна l- Клинодактилия 5 -го пальца l (искривлённый мизинец) — 60 %; l- Аркообразное ( «готическое» ) l нёбо — 58 %; l- Плоская переносица — 52 %; l- Бороздчатый язык — 50 %.
 — l")
Клиника синдрома Дауна l- Пигментные пятна по краю l радужки (пятна Брушфильда) — l 19 %; l- Эписиндром — 8 %; l- Стеноз или атрезия 12 -перстной l кишки — 8 %; l- Врождённый лейкоз — 8 %.

Формы синдрома Дауна l Примерно в 91 % случаев возникает ненаследственный вариант болезни — простая полная трисомия 21 хромосомы, обусловленная нерасхождением хромосом во время мейоза. Примерно у 5 % больных наблюдается мозаицизм (не все клетки содержат лишнюю хромосому). l В остальных случаях синдром вызван спорадической или наследуемой транслокацией 21 -й хромосомы. Как правило, такие транслокации возникают в результате слияния центромеры 21 -й хромосомы и другой акроцентрической хромосомы. l Фенотип больных определяется трисомией 21 q 22. l Повторный риск рождения ребёнка с синдромом Дауна у родителей с нормальным кариотипом составляет около 1 % при обычной трисомии у ребёнка.

Диагностика синдрома l Беременная женщина может пройти обследование на выявление нарушений плода. Многие стандартные дородовые обследования способны обнаружить синдром Дауна у плода. Например. имеются специфические УЗИ-признаки синдрома. l Генетические консультации с генетическими тестами (амниоцентез, биопсия хориона, кордоцентез), как правило, предлагаются семьям, риск рождения в которых ребёнка с синдромом Дауна наиболее велик. l l В США инвазивные и неинвазивные обследования доступны для всех женщин, вне зависимости от их возраста. l Однако инвазивные обследования проводить не рекомендуется, если женщине больше 34 -х лет и неинвазивные обследования не показали вероятных нарушений.

Диагностика синдрома l Амниоцентез и биопсия хориона считаются инвазивными обследованиями, так как при них в матку женщины вводят различные инструменты, что несёт в себе некоторый риск повреждения стенки матки, плода или даже выкидыша. l Риск выкидыша при биопсии хориона — 1 %, при амниоцентезе — 0, 5 %. l Существует несколько неинвазивных обследований, они, как правило, проводятся в конце 1 -го или в начале 2 -го триместра. l В каждом из них есть шанс получить ложноположительный результат, то есть обследование покажет, что у плода синдром Дауна, хотя на самом деле он здоров.

Диагностика синдрома l Даже с самыми лучшими обследованиями вероятность обнаружения синдрома составляет 90— 95 %, а уровень ложноположительных результатов 2— 5 %. l На данный момент аминоцентез считается самым точным обследованием. l Для получения результатов у женщины требуется взять на анализ амниотическую жидкость, в которой позже выявляют клетки плода. l Лабораторные работы могут занять несколько недель, но вероятность правильного результата — 99, 8 %. l Ложноположительный показатель очень низок.

БЛАГОДАРЮ ЗА ВНИМАНИЕ!
Генные и хром. заболвания.ppt