Генетика человека.ppt.pptx
- Количество слайдов: 37



Генетика человека 1. Особенности человека, как объекта исследования 2. Методы изучения наследственности человека 3. Наследственные болезни человека
 • Медленная смена")
Особенности человека, как объекта исследования • Невозможность искусственного скрещивания (мораль, этика) • Медленная смена поколений, мало потомков • Большое число групп сцепления(23 ♀ , 24♂) • Высокая степень полиморфизма (Полиморфизм -различия между гомологичными хромосомами, не оказывающие влияния на фенотип. Обеспечивает уникальность кариотипа каждого человека. ) • Невозможность создания одинаковых условий жизни.
 • Цитологический (кариотипирование) •")
Методы изучения наследственности человека • Клинико-генеалогический (составление и анализ родословных) • Цитологический (кариотипирование) • Близнецовый • Дерматоглифический • Биохимический • Метод гибридизации соматических клеток • Метод рекомбинантной ДНК (анализ и клонирование ДНК, создание геномных библиотек) • Популяционно-статистический • Метод моделирования (создание трансгенных животных моделирующих болезни человека)

Близнецовый метод Изучение закономерностей наследования признаков у моно- и дизиготных близнецов Позволяет: • Оценить степень влияния среды и генотипа на развитие признака в норме и патологии • Выявить наследственный характер признака • Определить пенетрантность аллеля • Оценить эффективность действия на организм некоторых внешних факторов (лекарств, обучения, воспитания) • Процент сходства группы близнецов по изучаемому признаку КОНКОРДАНТНОСТЬ; процент различия – ДИСКОРДАНТНОСТЬ • Сравнение конкордантности по данному признаку у монозиготных и дизиготных близнецов помогает более объективно судить о роли генотипа в формировании признака. ))).

Близнецовый метод Для оценки наследственности и среды в развитии признака используют формулу Хольцингера: Смб (%) – Сдб(%) Н= 100% - Сдб (%) Н- коэффициент наследственности Смб – конкордантность монозиготных близнецов Сдб – конкордантность дизиготных близнецов Н=1 →признак определяется генотипом Н=0 → признак определяется средой 0<Н<1 →влияние и среды и наследственности

Клинико-генеалогический метод • Составление и анализ родословных • Выявление типа и характера наследования признака : Ø аутосомно-доминантное, Ø аутосомно-рецессивное, Ø сцепленное с полом, Ø Нетрадиционные типы наследования: цитоплазматическое наследование, однородительские дисомии, болезни экспансии) • Определение пенетрантности и экспрессивности гена • Вычисление генетического риска
 Сбор сведений о семье(анамнез) § (прежде всего сбор сведений")
Клинико-генеалогический метод. СОСТАВЛЕНИЕ РОДОСЛОВНОЙ: 1) Сбор сведений о семье(анамнез) § (прежде всего сбор сведений о пробанде — индивиде, который является предметом интереса исследователя (врача, педагога). Чаще всего это больной или носитель изучаемого признака. )) § Дети одной родительской пары (братья и сестры) называются сибсами. § Если сибсы имеют только одного родителя, они называются полусибсами. Различают: единоутробных сибсов (общая мать); единокровных сибсов (общий отец). § Обычно родословная собирается в связи с изучением одного или нескольких заболеваний (признаков). Чем больше поколений вовлекается в родословную, тем больше информации она может содержать. § Для составления родословной используют определенные символы: 2) Построение родословной 3) Анализ родословной и выводы


Родословная с концентрическим расположением поколений
")
Родословная семьи с брахидактилией (недоразвитость дистальных фаланг пальцев, приводящая к короткопалости)

Цитологический метод • Кариотипирование – анализ числа и морфологии хромосом клетки на стадии метафазы митоза (определение пола, диагностика хромосомных болезней, изучение мутагенеза), составление кариограммы и идеограммы. • Изучение полового хроматина (экспресс-диагностика половой принадлежности, быстрое определение хромосомных болезней, связанных с изменением числа хромосом по количеству телец Барра) • Метод флуоресцентной in situ гибридизации (FISH): локализация генов в хромосомах, выявление хромосомных аномалий при пренатальной диагностике, ген. Тестирование эмбрионов при ЭКО)

Тельце Барра

Кариограмма человека. А- женщины, Б-мужчины

Идеограмма нормального кариотипа человека
 Ø размеры хромосом, Ø")
Анализ кариограммы Денверская классификация хромосом (1960 -1966 г. г. ) Ø размеры хромосом, Ø положение центромеры
 Ø размеры хромосом Ø положение центромеры Ø дифференциальное")
Анализ кариограммы Парижская классификация (1971 год) Ø размеры хромосом Ø положение центромеры Ø дифференциальное окрашивание Парижская классификация хромосом (1971 г)
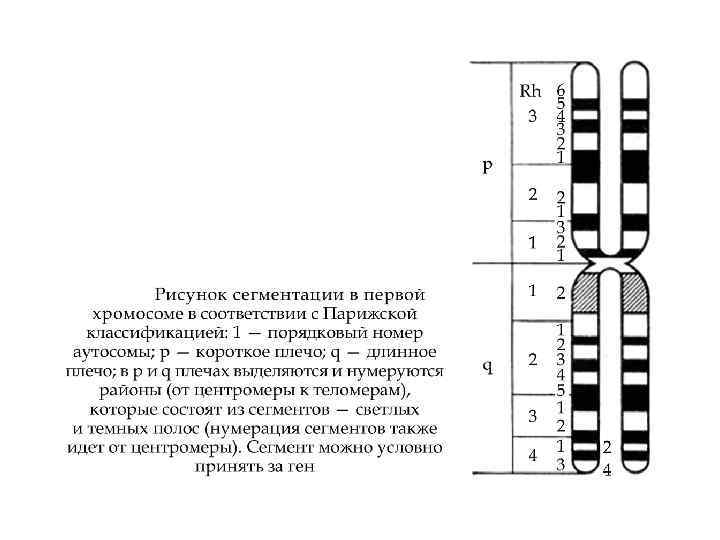
")
Метод флуоресцентной in situ гибридизации (FISH)
 Метафазная пластинка и интерфазные ядра из лимфоцитов пациента")
Метод флуоресцентной in situ гибридизации (FISH) Метафазная пластинка и интерфазные ядра из лимфоцитов пациента с кариотипом 47, ХХУ после 5 -цветной флуоресцентной гибридизации in situ. Участки хромосом маркированы цветами: 13 хр. – красным, 21 хр. - зеленым, 18 хр. - голубым, Х хр. -синим, Yхр. - желтым

Дерматоглифический метод • Изучение рисунка на ладонях, подошвах и пальцах. (дактилоскопия - пальцы, пальмоскопия - ладони, плантоскопия – ступни ног ) • Используется в медико-генетических консультациях для уточнения клинического диагноза, • дифференциации различных форм заболеваний, • для выяснения носителей мутантных аллелей. Ø Синдром Дауна (Трисомия 21) - наличие «Обезьяньей линии» на обеих руках, учащение узоров на подушечке под большим пальцем. Ø Синдром Клайнфельтера (47, ХХУ) - учащенный узор на подушечке под большим пальцем Ø изменение дерматоглифики обнаружены при множественных, врожденных пороках развития ЦНС, сердечно – сосудистой системы, желудочно-кишечного тракта.

Биохимический метод • Обнаружение биологически активных соединений и их метаболитов в жидкостях организма • Позволяет выявить нарушения в обмене веществ, вызванные изменением генов и, как следствие, изменением активности различных ферментов. (фенилкетонурия, серповидно-клеточная анемия) С помощью биохимических методов открыто около 500 молекулярных болезней, являющихся следствием проявления мутантных генов. К наследственным болезням обмена веществ относят болезни углеводного обмена (сахарный диабет), обмена аминокислот, липидов, минералов и др. . • Биохимический скрининг по определению содержания альфафетопртеина (АФП) и хорионического гонадотропина (ХГ) в крови беременных на сроке 1518 недель с целью выявления женщин, имеющих повышенный риск рождения ребенка с хромосомными болезнями (синдром Дауна и Эдвардса), а так же с ДЗНТ (дефекты заращения нервной трубки). • При синдроме Дауна у плода уровень АФП в крови беременной ниже в 2 и более раз. При наличии синдрома Эдвардса у плода резко снижен уровень ХГ. При анэнцефалии, открытой spina bifida, дефектах передней брюшной стенки (омфалоцеле, гастрошизисе, гидронефрозе и т. д. ) уровень АФП в 3 и более раз повышен по сравнению с нормой. • Трудоемкость • Требуется специальное оборудование

Метод гибридизации соматических клеток • основан на размножении соматических клеток в искусственных условиях • используются культуры соматических клеток, полученные из материала биопсий (кровь, кожа, опухолевая ткань, ткань эмбриона) для генетических исследований человека. • дает возможность анализировать генетические процессы в отдельных клетках и использовать их для изучения генетических закономерностей целостного организма. • позволяет изучать механизмы первичного действия и взаимодействия генов, регуляцию генной активности.
")
Популяционно-статистический метод Позволяет изучить: • распространение отдельных генов и признаков(в том числе и заболеваний) в человеческих популяциях, • соотношение между частотой гомозигот и гетерозигот • роль наследственности и среды в возникновении болезней, особенно с наследственной предрасположенностью. Производится: выборочное исследование части популяции: изучение архивов больниц, родильных домов, проводится опрос путем анкетирования Используются: методы статистического анализа (закон Г. Харди и В. Вайнберга и ряд других специальных математических методов) для определения частоты генов в различных группах населения, частоты гетерозиготных носителей ряда наследственных аномалий и болезней. Например, гомозиготы по гену Нb. S в Беларуссии практически не встречаются, а в странах Западной Африки частота их варьирует от 25% в Камеруне до 40% в Танзании. Изучение распространения генов среди населения различных географических зон (геногеография) дает возможность установить центры происхождения различных этнических групп и их миграции, определить степень риска появления наследственных болезней у отдельных индивидуумов.
 Различные клинические проявления Минимальное")
• • • Разнообразие (различные мутации в одном гене) Различные клинические проявления Минимальное влияние внешней среды ГЕННЫЕ Ø Аминокислотный обмен (альбинизм, фенилкетонурия) Ø Углеводный обмен (болезнь Тэя-Сакса) Ø Гемоглобинопатии Ø Дефекты биосинтеза гормонов (адреногенитальный синдром, синдром тестикулярной феминизации) Ø Мышечные дистрофии, муковисцидоз Патологические реакции на факторы внешней среды: Лекарства, пищевые добавки, тепло, холод, солнечный свет… Болезни предрасположенности (псориаз, шизофрения, сердечно-сосудистые з-я…) МУЛЬТИФАКТОРИАЛЬНЫЕ Наследственные болезни человека ХРОМОСОМНЫЕ Геномные гетероплоидии: Структурные изменения: Полиплоидии (3 n=69, 4 n=92) Внутрихромосомныеделеции, инверсии, дупликации… Анеуплоидии – Межхромосомные транслокации- моносомии (2 n-1=45) Сбалансированные(реципрокные) нуллисомии (2 n-2=44), Несбалансированные (нереципрокные) трисомии (2 n+1=47), полисомии(2 n+2=48, 2 n+3=49 Робертсоновские транслокации для Х, Y) БОЛЕЗНИ С НЕТРАДИЦИОННЫМ ТИПОМ НАСЛЕДОВАНИЯ Ø Митохондриальные болезни Ø Болезни экспансии (хорея Гентингтона, с-м ломкой хромосомы=с-м Мартина-Белл) Ø Генный импринтинг (синдром Прадера-Вилли, 15 Хр. )

Хромосомные болезни человека Нормальный кариотип - 46, ХХ женщина; 46, ХY мужчина Изменение числа аутосом (аутосомные мутации) Синдром Дауна – 47, ХХ+21, 47, ХY+21 Синдром Эдвардса – 47, ХХ+18; 47, ХY+18 Синдром Патау - 47, ХХ+13; 47, ХY+13 Изменение числа половых хромосом (генеративные мутации) Синдром Шерешевского-Тернера – 45, ХO Синдром Клайнфельтера- 47, ХХY ( 48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY; 49 XXXYY) Полисомия Х хромосомы. Трисомия по Х -47, ХХХ (48, ХХХХ; 49, ХХХХХ…) Структурные перестройки Хронический миелолейкоз-46, ХХ, t(9/22); 46, ХY, t(9/22)-реципрокная перестройка Транслокационный С. Дауна-46, ХХ, t(15/21); 46, ХY, t(15/21) – Робертсоновская транслокация Синдром Орбели - 46, ХХ, del(13 q-); 46, ХY, del(13 q-) делеции Синдром «Кошачьего крика» синдром Лежёна -46, ХХ, del(5 p-); 46, ХY, del(5 p-)
 обусловлен делецией длинного плеча 13 -й хромосомы, сегментов 13 q")
Синдром Орбели (13 q-) обусловлен делецией длинного плеча 13 -й хромосомы, сегментов 13 q 22 -q 31. Популяционная частота синдрома не установлена. Дети с синдромом Орбели рождаются с низким весом (2200 г). Клинически синдром проявляется аномалиями развития всех систем организма. Характерны микроцефалия, отсутствие носовой вырезки (лоб непосредственно переходит в нос), эпикант, антимонголоидный разрез глаз, широкая спинка носа, высокое нёбо, низко расположенные деформированные ушные раковины. Отмечаются поражения: • глаз (микрофтальмия, иногда анофтальмия, косоглазие, катаракта, ретинобластома), • опорно-двигательного аппарата (короткая шея, гипо- или аплазия первого пальца кисти и пяточной кости, синдактилии кистей и стоп), • атрезии прямой кишки и заднепроходного отверстия. • Часты пороки развития сердца, почек, головного мозга. Для всех детей с синдромом Орбели характерна глубокая олигофрения, возможны потеря сознания, судороги. Большинство больных с синдромом 13 q- погибают на 1 -м году жизни.
 обусловлен делецией короткого плеча 5 -й хромосомы. Популяционная частота")
Синдром кошачьего крика (5 р-) обусловлен делецией короткого плеча 5 -й хромосомы. Популяционная частота синдрома -примерно 1: 45 000. Для данного синдрома наиболее характерны: • специфический плач, напоминающий кошачье мяуканье, • лунообразное лицо, • мышечная гипотония, • умственное и физическое недоразвитие, микроцефалия, • низко расположенные, иногда деформированные ушные раковины, эпикант, антимонголоидный разрез глазных щелей, косоглазие. Иногда наблюдаются атрофия зрительного нерва и очаги депигментации сетчатки. Как правило, выявляются пороки сердца. Наиболее постоянный признак синдрома - "кошачий крик" - обусловлен изменениями гортани: сужением, мягкостью хрящей, отечностью или необычной складчатостью слизистой оболочки, уменьшением надгортанника. Изменения других органов и систем неспецифичны. Продолжительность жизни у больных с этим синдромом значительно снижена, только около 14% из них переживают возраст 10 лет.
; 46, ХY, t(9/22) (хронический миелобластный лейкоз, хронический миелолейкоз, ХМЛ) хромосомная")
Хронический миелолейкоз-46, ХХ, t(9/22); 46, ХY, t(9/22) (хронический миелобластный лейкоз, хронический миелолейкоз, ХМЛ) хромосомная транслокация, приводящая к образованию филадельфийской хромосомы, получившей своё название по месту работы её первооткрывателей, Питера Ноуелла и Дэвида Хангерфорда, 1960 г. Филадельфиия) • болезнь, при которой наблюдается избыточное образование гранулоцитов в костном мозге и повышенное накопление в крови как самих этих клеток, так и их предшественников. Слово «хронический» в названии болезни означает, что процесс развивается сравнительно медленно, в отличие от острого лейкоза, а «миелоидный» означает, что в процесс вовлечены клетки миелоидной (а не лимфоидной) линии кроветворения.
 1866 г. , Джон Даун, Англия, педиатр; 1959 г.")
Синдром Дауна (47, ХХ/XY +21) 1866 г. , Джон Даун, Англия, педиатр; 1959 г. Жером Лежен, Франция, генетик –хромосомная природа 1: 700 -800 новорожденных Причины: • Трисомия по 21 хромосоме (нерасхождение в І делении Мейоза, риск повышается с возрастом родителей; ~ 94%) • Робертсоновские транслокации 14/21; 15/21 хромосом (семейная форма, транслокация возникает у родителей, не отражаясь на фенотипе, возраст родителей не имеет значения; ~4%) • Мозаицизм (нерасхождение возникает в клетке зародыша на ранних стадиях его развития, нарушение кариотипа затрагивает только некоторые ткани и органы (46, XX/47, XX, +21); ~2%)
 § монголоидный")
Симптомы: § плоское лицо, § плоский затылок § брахицефалия (аномальное укорочение черепа) § монголоидный разрез глаз, § эпикантус (вертикальная кожная складка, прикрывающая медиальный угол глазной щели) § гиперподвижность суставов § поперечная ладонная складка «обезьянья» § брахимезофалангия (укорочение всех пальцев за счёт недоразвития средних фаланг) § § слабый тонус мышц повышенное содержание пуринов когнитивные нарушения раннее старение Сопутствующие заболевания: • врожденные пороки сердца, • болезнь Альцгеймера, • острый миелоидный лейкоз • ослабленный иммунитет • нарушения пищеварения Широкая посадка между большим и следующим пальцем ноги
 Причины: трисомия по хромосоме 18 Джон Эдвардс, Англия, цитогенетик, 1960 г.")
Синдром Эдвардса(47, ХХ/ХY+18) Причины: трисомия по хромосоме 18 Джон Эдвардс, Англия, цитогенетик, 1960 г. 1: 7000 новорожденных, ♀ в 3 раза больше ♂ • Трисомия по 18 хромосоме (нерасхождение хромосом в Мейозе, риск повышается с возрастом матери; ~ 90%) • Мозаицизм (нерасхождение возникает в клетке зародыша на ранних стадиях его развития, нарушение кариотипа затрагивает только некоторые ткани и органы (46, XX/47, XX, +18); ~10%) Симптомы: § Низкая масса при рождении § Сниженная двигательная активность § § § аномалии мозгового и лицевого черепа глазные щели узкие и короткие ушные раковины деформированы аномальное развитие стопы пороки сердца и крупных сосудов Продолжительность жизни : 60 % детей доживают до 3 месяцев, до года 5 -10 %. Основная причина смерти - остановка дыхания и нарушения работы сердца и почек. Оставшиеся в живых — глубокие олигофрены
 трисомия по хромосоме 13 Клаус Патау, американский генетик, 1960 1:")
Синдром Патау(47, ХХ/ХY +13) трисомия по хромосоме 13 Клаус Патау, американский генетик, 1960 1: 6000 -9000 новорожденных, ♀=♂ Причины: • Трисомия по 13 хромосоме (нерасхождение хромосом в Мейозе, риск повышается с возрастом матери; ~ 85%) • Робертсоновские транслокации 13/14 хромосом • Мозаицизм (нерасхождение возникает в клетке зародыша на ранних стадиях его развития, нарушение кариотипа затрагивает только некоторые ткани и органы (46, XX/47, XX, +13); редко) Симптомы: § Низкая масса при рождении § Сниженная двигательная активность § § § аномалии мозгового и лицевого черепа Скошенный лоб Запавшая переносица Расщелины губы и неба полидактилия пороки сердца и крупных сосудов § изменения поджелудочной железы, добавочные селезёнки, § пороки развития половых органов Продолжительность жизни : 95% детей доживают до года, (до 10 лет 2 — 3 % детей). Оставшиеся в живых — глубокие олигофрены
 полисомия по половым хромосомам Гарри Клайнфельтер, американский врач, 1942 г.")
Синдром Клайнфельтера (47, ХХY) полисомия по половым хромосомам Гарри Клайнфельтер, американский врач, 1942 г. 1: 500 -600 новорожденных мальчиков Варианты: 48 ХХХY, 49 ХХХYY…. Причины: патология мейоза - нерасхождение хромосом при овогенезе ~70%, при сперматогенезе ~30% Факторы риска - возраст матери Клинические проявления § диспропорциональное строение тела – высокая талия, длинные конечности § Гинекомастия (увеличение грудных желез) § ожирение и оволосение по женскому типу § недоразвитие сперматозоидов, бесплодие § Психологические особенности- манерность, болтливость § Умственное развитие-от нормы до легкой степени УО (? )

 1925 г. , Николай Адольфович Шерешевский, СССР, эндокринолог; 1938 г. ,")
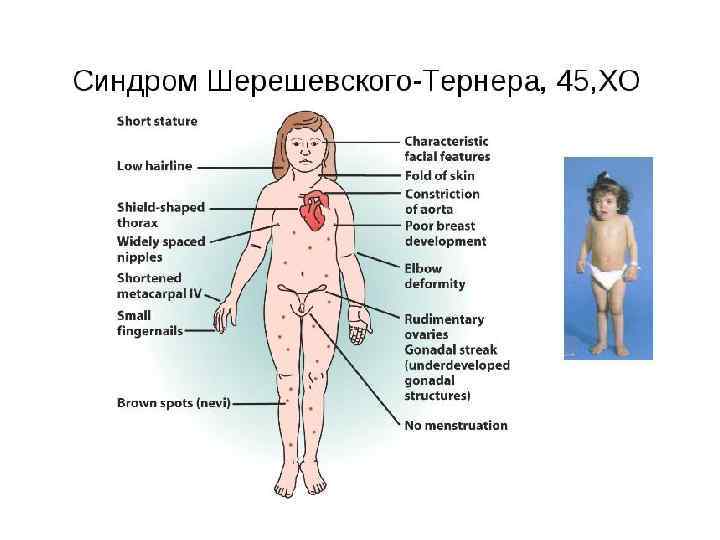
Синдром Шерешевского-Тернера(45, ХО) 1925 г. , Николай Адольфович Шерешевский, СССР, эндокринолог; 1938 г. , Генри Тёрнер, США - врач – триада симптомов 1959 г. , Ч. Форд – раскрыта этиология заболевания (моносомия по Xхромосоме) 1: 3000 девочек Причины: нарушения расхождения хромосом при гаметогенезе Симптомы: • половой инфантилизм • кожные крыловидные складки на боковых поверхностях шеи • деформация локтевых суставов • Низкий рост (135 -145 см) • Интеллект-норма (характерен психический инфантилизм с эйфорией)

n+1 n-1
Генетика человека.ppt.pptx