bf3825b266c127165f4f06d3032e2493.ppt
- Количество слайдов: 78



Геморрагические заболевания у детей Лекция для студентов VI курса Доцент Кантемирова М. Г.
, а")
Вопрос 1. Укажите, какие тесты на кровоточивость диагностируют нарушения сосудисто-тромбоцитарного механизма гемостаза (А), а какие – нарушения коагуляционного гемостаза (Б)? 1. 2. 3. 4. 5. 6. 7. 8. Определение количества тромбоцитов Изучение мазка периферической крови Время кровотечения Активированное частичное тромбопластиновое время Протромбиновое время МНО Тромбиновое время Концентрация фибриногена в плазме
, а какие для болезни Верльгофа(Б)? Ø")
Вопрос 2. Какие клинические проявления характерны для гемофилии(А), а какие для болезни Верльгофа(Б)? Ø Ø Ø 1. Длительные отсроченные кровотечения 2. Спонтанные носовые, десневые, желудочнокишечные кровотечения 3. Кожный геморрагический синдром: экхимозы и петехии 4. Полихромные, несимметричные кожные геморрагии 5. Подкожные, межмышечные, периневральные гематомы 6. Гемартрозы
: клинико-гематологический синдромы, объединяющие различные по этиологии и патогенезу заболевания, отличительным и")
Геморрагические заболевания (диатезы): клинико-гематологический синдромы, объединяющие различные по этиологии и патогенезу заболевания, отличительным и главным общим признаком которых является повышенная патологическая кровоточивость.

Гемостаз – функциональная система организма, обеспечивающая, с одной стороны, остановку и предупреждение кровотечений при нарушении целостности сосудистой стенки, а с другой – сохранение жидкого состояния циркулирующей и депонированной крови.

3 звена гемостаза: звено Сосудистое Тромбоцитарное звено Плазменное звено

Механизмы гемостаза 1. Сосудисто-тромбоцитарный - первичный 2. Коагуляционный - вторичный l l Внутренний Внешний

Гемостатическая функция эндотелия и субэндотелия микрососудов Рефлекторный спазм микрососудов. Ø Секреция катехоламинов, серотонина, АДФ – поддержание спазма и стимуляция агрегации Тр. Ø Выделение в кровь активированных факторов гемостаза: тканевой тромбопластин (III ф. ), ф. Виллебранда, простациклин. Ø Контактная активация коллагеном адгезии и агрегации Тр. и XII ф. Ø

Гемостатическая функция тромбоцитов Ø Ангиотрофическая ф-ция (поддерживает нормальную структуру и функцию эндотелия, утойчивость к повреждени) Ø Секреция вазоактивных в-в (катехоламинов, серотонина, АДФ и др. ) Ø Формирование первичного тромба за счет адгезии и агрегации Тр.

Cхема первичного сосудистоготромбоцитарного гемостаза Повреждение микрососудов Синтез ф. Виллебранда Выделение тканевого тромбопластина Синтез ГП Iв Обнажение коллагена субэндотелия Выделение в кровь Локальный серотонина спазм катехоламинов, микрососудов АДФ и др. Контактная активация тромбоцитов и XII ф. Адгезия тромбоцитов Синтез тромбоксана Агрегация тромбоцитов Первичный тромбоцитарный тромб

Коагуляционный гемостаз - это сложный каскадный ферментативный процесс свертывания крови, имеющий внешний и внутренний механизм активации и характеризующийся двумя основными фазами: - ферментативный процесс активации протромбина (ф. II) в тромбин (ф. IIа) - превращение фибриногена в фибрин под влиянием тромбина

Факторы свертывания крови Фактор II – протромбин Ø Фактор IV – ионизированный кальций Ø Фактор V - проакцелирин, Ас-глобулин Ø Фактор VII – проконвертин, антифибринолизин Ø Фактор VIII – антигемофил. глобулин А Ø Фактор Х – ф. Стюарта-Прауэр Ø Фактор XI – агтигемоф. глобулин С Ø Фактор XII – контактный ф. Хагемана Ø


Система фибринолиза – неотъемлимая часть гемостаза Ø Направлена на сохранение жидкого состояния крови, препятствует переходу локального тромбооброзования в распространенное.

Скрининг-тесты на кровоточивость Определение количества тромбоцитов 2. Изучение мазка периферической крови 3. Время кровотечения < 4 мин. 4. Активированное частичное тромбопластиновое время 29 -34 с 5. Протромбиновое время 9, 2 -12, 2 с 6. МНО 1, 5 -2 (на фоне варфарина 2, 5 -3, 0) 7. Тромбиновое время 18 -24 с 8. Концентрация фибриногена в плазме Дополнительные пробы Определение протеина С, S, антитромбина III Ø Определение ПДФ или уровня D-димера Ø Определение содерж. факторов свертывания крови 1.
 Ø Васкулитно-пурпурный Ø Смешанный микроциркуляторно- гематомный")
Пять типов кровоточивости: Ø Гематомный Ø Петехиально-пятнистый (синячковый) Ø Васкулитно-пурпурный Ø Смешанный микроциркуляторно- гематомный Ø Ангиоматозный

Гематомный тип Массивные, глубокие, напряженные, болезненные кровоизлияния в крупные суставы, мышцы, подкожную клетчатку, апоневрозы, фасции, серозные оболочки. Ø Спонтанные, посттравматические, постоперационные кровотечения, имеющие отсроченный характер. Ø Характерен для нарушений внутреннего механизма протромбиназной активности (гемофилия А и В, ингибиторные формы). Ø

Гемартроз у больного гемофилией

Петехиально-пятнистый тип Ø Ø Ø Безболезненные, не напряженные, не сдавливающие окружающие ткани, не вызывающие их деструкции кровоизлияния в кожу и слизистые оболочки (петехии, экхимозы). Десневые, носовые, маточные кровотечения. Кровотечения возникают при ничтожно малой травматизации микрососудов. Отмечаются длительные кровотечения после удаления зубов. Наблюдается при тромбоцитопении, тромбоцитопатиях, а также при гипо- и дисфибриногенемиях, дефиците факторов X, V, II.

Васкулитно-пурпурный тип Ø Характеризуется геморрагиями в виде сыпи или эритемы на воспалительной основе. Ø Возможно присоединение нефрита и кишечных кровотечений. Ø Характерно наличие суставного синдрома, субфебрилитет. Ø Наблюдается при васкулитах.

Геморрагические диатезы Коагулопатии Тромбоцитопатии и тромбоцитопении Вазопатии Геморрагические гематомезенхимальные дисплазии

ГЕМОРРАГИЧЕСКИЕ МЕЗЕНХИМАЛЬНЫЕ ДИСПЛАЗИИ Ø Группа геморрагических диатезов, обусловленная патологией развития соединительной ткани (в большей степени — коллагена), а также нарушением различных компонентов гемостаза (сосудистого, тромбоцитарного, плазменного).

Этиология Ø Наследственная патология. К геморрагическим мезенхимальным дисплазиям относятся геморрагические варианты синдрома Элерса-Данло, синдрома Марфана, Тар-синдрома (тромбоцитопатия и тромбоцитопения в сочетании с врожденным отсутствием лучевых костей), Тар-синдрома с недостаточностью факторов VII или X

Клиника Ø Наблюдаются геморрагический синдром и проявления соответствующих сосудистых и мезенхимальных нарушений. Отмечаются дефекты развития костной ткани, связочного аппарата, гипермобильность суставов, повышенная растяжимость кожи, пролабирование клапанов сердца.

Коагулопатии у детей.
 Гемофилия В")
Коагулопатии Наследственные Ø Ø Ø Гемофилия А ( дефицит VIII ф. ) Гемофилия В ( дефицит IX ф. ) Болезнь Виллебранда Наследственный дефицит факторов XI (гемофилия С) XII, XIII и I ф. Дисфибриногенемии Наследственный дефицит факторов VII, X, V и II Приобретенные Дефицит Квитаминозависимых факторов свертывания Ø ДВС-синдром Ø Синдром массивных трансфузий Ø Нарушения гемостаза, вызванные неспецифическими иммунными ингибиторами – антикоагулянтами волчаночного типа (АВТ) Ø

Наследственные коагулопатии с геморрагическими проявлениями
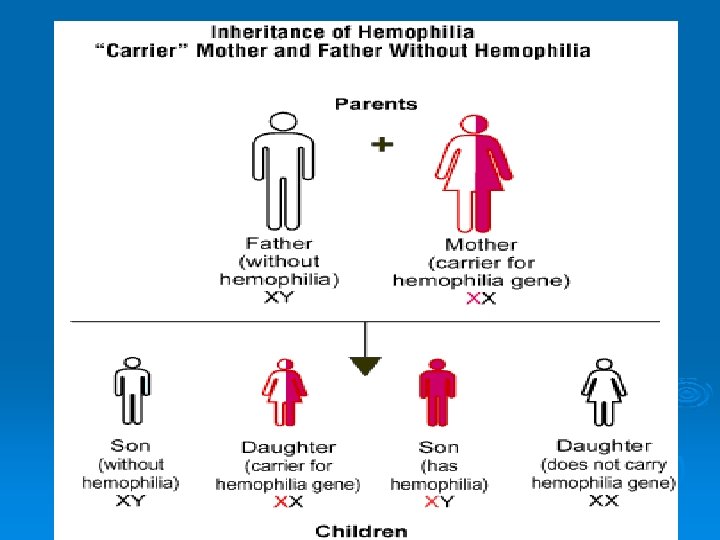

Наследственные коагулопатии с геморрагическими проявлениями Гемофилия Ø Ø Ø Ø Гематомный тип кровоточивости Длительные, иногда спонтанные кровотечения отсроченного характера В неонатальном периоде может манифестировать с кефалогематомы, внутричерепных геморрагий, кровоизлияния в области ягодиц при ягодичном предлежании, кровотечения из пупочной ранки, уздечки языка при плаче; После года, с активизацией двигательного режима - гемартрозы и межмышечные гематомы, кровотечения из лунок зубов, со слизистой полости рта, кровотечения при циркумцизии, на месте внутримышечных и внутрикожных инъекций; относительно редко наблюдаются носовые кровотечения и геморрагии внутренних органов. Внутрибрюшные гематомы, забрюшинные гематомы, возникающие в возрасте 2 -3 лет, характеризуются повышенной болезненностью, приводят к компрессии окружающих тканей, нарушая функцию органов и костей (костная псевдоопухоль). Кровоизлияние в подвздошно-поясничную мышцу – ригидность мышц передней брюшной стенки ( «поражение тазобедренного сустава» , «острый аппендицит» );

Ø Ø Гематурия – чаще у детей старше 5 лет. Причины: травмы поясничной области, частый прием анальгетиков, иммунокомплексное поражение почек, оксалурия у больных с повторными гемартрозами. Может сопровождаться дизурией, болями в поясничной области (вплоть до почечной колики). Желудочно-кишечные кровотечения характерны для детей старшего возраста, осложняют эрозивноязвенную патологию. Кровоизлияния в головной мозг - стремительное развитие (беспокойство или заторможенность, рвота, головная боль, стволовые симптомы: нистагм, анизокория, расстройства ритма дыхания и сердечных сокращений), в спинной мозг – нарастающие периферические парезы. Заместительная терапия. Легкая форма может проявиться лишь в зрелом возрасте при оперативных вмешательствах и значительных травмах.

Наследственные коагулопатии с геморрагическими проявлениями

Severity I. Individuals with less than 1% active factor are classified as having severe haemophilia, II. Individuals with 1 -5% active factor have moderate haemophilia III. Individuals with 5 -40% of normal levels of active clotting factor - mild haemophilia

Лабораторная диагностика гемофилии Удлинение времени свертывания цельной крови и АЧТВ Ø Нормальные время кровотечения и ПТВ Ø Тип и тяжесть – на основании степени снижения коагулянтной активности антигемофильных глобулинов в плазме Ø Нормальное содержание антигена ФВ Ø Пренатальная диагностика: ПЦР (биоптат ворсинок хориона, кровь плода) уже на сроке 8 -12 недель Ø

Laboratory findings in various platelet and coagulation disorders Partial Prothrombin thromboplastin Condition time Normal or Vitamin K mildly Prolonged deficiency or warfarin prolonged Disseminated intravascular Prolonged coagulation Bleeding time Platelet count Unaffected Prolonged Decreased Von Willebrand disease Unaffected Prolonged Hemophilia Aspirin Thrombocytopenia Liver failure, early Unaffected Unaffected Prolonged Unaffected Decreased and/or Rejected Unaffected Decreased Unaffected Liver failure, end-stage Uremia Prolonged Unaffected Prolonged Decreased Unaffected Congenital afibrinogenemia Prolonged Unaffected Factor V deficiency Prolonged Unaffected Factor X deficiency as seen in amyloid purpura Prolonged Unaffected Glanzmann's thrombasthenia Unaffected Prolonged Unaffected Bernard-Soulier syndrome Unaffected Prolonged Factor XII deficiency Unaffected Prolonged Unaffected Decreased or unaffected Unaffected

Лечение гемофилии Ø I. Ср-ва, влияющие на свертывающую систему крови: 1. очищенные и рекомбинантные концентраты антигемофильных глобулинов (АГГ) 2. антифибринолитики (аминокапроновая, аминометилбензойная, транексамовая к-ты)

Принципы лечения концентратами АГГ при гемофилии 1. АГГ применяют при кровотечениях Ø 2. перед хирургическими вмешательствами Ø 3. АГГ при непрерывной профилактике кровотечений (у детей 2 -3 раза в неделю) Ø 4. доза АГГ зависит от локализации кровотечения, его прогнозе, исходного уровня активности VIII, XI ф и веса пациента Ø
 Ø II. Местная терапия (фибриновый клей, гемостатическая губка, фибриновая пленка). Ø")
Лечение гемофилии (продолжение) Ø II. Местная терапия (фибриновый клей, гемостатическая губка, фибриновая пленка). Ø III. Сопутствующая терапия (при гемартрозах –иммобилизация сустава, анальгетики, в/с пункция с аспирацией крови и введением гидрокортизона; преднизолон при почечных кровотечениях; дегидратация, противосудорожные препараты при в/ч кровоизлияниях)

Болезнь Виллебранда — наследственное заболевание крови, характеризующееся возникновением эпизодических спонтанных кровотечений, которые схожи с кровотечениями при гемофилии. Заболевание наследуется по принципу аутосомного доминирования. Причина кровотечений — нарушение свертываемости крови из-за недостаточной активности фактора Виллебранда. Распространенность болезни Виллебранда составляет 1 на 800— 1000.

Болезнь Виллебранда Ø Болезнь фон Виллебранда названа в честь Адольфа Эрика фон Виллебранда, финского педиатра, который впервые описал это заболевание в 1926 году.

Адольф Эрик фон Виллебранд
 в условиях")
Функции фактора Виллебранда 1. Опосредование адгезии тромбоцитов к коллагену субэндотелия (и аггрегации) в условиях высокой скорости тока крови 2. Связывание фактора VIII: Ø– Зашита от преждевременной протеолитической инактивации – Доставка и создание высокой концентрации в области повреждения Активность пропорциональна молекулярной массе (чем больше размеры мультимера, тем он активнее). Синтезируется эндотелиальными клетками и мегакариоцитами

» Фактор Виллебраннда - это мультимерный гликопротеин, который необходим для адгезии тромбоцитов(прилипание, прикрепление тромбоцитов к сосудистой стенки в зоне повреждения целостности ). эндотелия

Наследственные коагулопатии с геморрагическими проявлениями Болезнь Виллебранда Ø Ø Ø Является следствием количественных или качественных нарушений ФВ. Бывает наследственной и приобретенной. Причина наследственной БВ – мутация гена, кодирующего синтез ФВ. Является самым распространенным геморрагическим заболеванием. Частота носительства дефектного гена 1: 100, но лишь 10 -30% носителей имеют клинические проявления. ФВ содержится в α-гранулах Тр, эндотелиоцитах, в плазме и субэндотелиальном матриксе. ФВ состоит из мультимеров с разной молекулярной массой, чем она больше, тем выше тромбогенный потенциал молекулы. Роль ФВ в гемостазе: опосредует адгезию Тр к субэндотелиальным структурам и между собой в процессе образования тромба, служит носителем VIIIф в плазме, удлиняя время его циркуляции.

Наследование БВ Ø На сегодня известно четыре типа наследственной болезни Виллебранда - тип 1, тип 2, тип 3, и тромбоцитарный тип. Международное общество по тромбоза и гемостаза (ISTH) классифицирует болезнь фон Виллебранда согласно определению качественного и количественного дефектов фактора фон Виллебранда.
) Тип 1")
Формы болезни фон Виллебранда Ø Ø (Международное общество тромбоза и гемостаза (ISTH)) Тип 1 (60 -80% всех случаев заболевания) - это количественное нарушение, недостаточный уровень ФВ Тип 2 (20 -30% всех случаев заболевания) - это качественное нарушение, , уровень фактора Виллебранда - нормальный, но структура нарушена Тип 3 обусловлен нарушением биосинтеза FW и характеризуется практически полным его отсутствием в плазме и тромбоцитах. Наследуется тип 3 по аутосомно-рецессивному. Тромбоцитарный тип (псевдоболезнь фон Виллебранда) - это аутосомно-доминантный тип БВ вызван усилением мутаций гена, кодирующего деятельность тромбоцитарного рецептора фактора Виллебранда. В данном случае мутация приводит к изменению участка гликопротеида Ib/IX, который связывает фактор фон Виллебранда. Уровни антигена фактора фон Виллебранда и фактора VIII нормальны. Активность ристоцетина и потеря больших мультимер Виллебранда, делает этот тип похожим на тип 2 B, но генетическое тестирование фактора Виллебранда не обнаружит никаких мутаций.

Учитывая аутосомно-доминантный тип наследования, генетический риск для потомства составляет 50 % независимо от пола плода. Ген, кодирующий деятельность ФВ расположен на двенадцатой хромосоме. Болезнь Виллебранда 1 и 2 типа, тромбоцитарный тип наследуются по аутосомнодоминантному типу, в то время как 3 тип наследуется по аутосомнорецессивному типу. Однако, известны случаи, когда 2 тип также наследовался рецессивно.

Ø Ø Ø Ø Ø Микроциркуляторный или смешанный тип кровоточивости; Характерны первичные кровотечения, начинающиеся сразу после травмы; Кожный геморрагический синдром: экхимозы, петехии; Кровотечения из травмированных слизистых оболочек, длительные кровотечения из лунок удаленных или выпавших зубов, рецидивирующие носовые кровотечения; Маточные кровотечения у девочек после начала менструаций; Интра- и послеоперационные кровотечения; Желудочно-кишечные кровотечения, Кровотечения из мочевых путей. Возможны кровотечения из мест инъекций и гематомы мягких тканей после различных травм

Клинические проявления БВ • Обильные носовые кровотечения 5% – 60% • Десневые кровотечения 7% - 51% • Выраженный кожный гемосиндром 12% - 24% (экхимозы, реже гематомы) • Кровотечения после удаления зубов 1% - 13% • Кровотечения после тонзилэктомии 2, 4% - 11% • Послеродовые кровотечения 6% - 23% • Меноррагии 23% - 44% • Гемартрозы ? • Внутричерепные кровоизлияния ? • После- и интраоперационные кровотечения? (слайд профессора П. В. Свирин) Ø Ø Ø

Приобретенная БВ Возникает на фоне сердечно-сосудистых, онкологических заболеваний и системных заболеваний соединительной ткани (стеноз аортального клапана, опухоль Вильмса, гипотиреоз, мезенхимальные дисплазии. Патогенез: ØСпецифические АТ к ФВ ØНеспецифические АТ – образование ИК и более активный клиренс ФВ ØАбсорбция ФВ клетками злокачественных опухолей ØСнижение синтеза или высвобождения ФВ ØПовышение протеолитической деградации ФВ

Диагностика Диагностические критерии болезни фон Виллебранда: Ø семейный анамнез, Ø смешанный тип кровоточивости, Ø увеличение времени кровотечения. Оценка фактора фон Виллебранда: Ø количественное содержание фактора фон Виллебранда (исследование ристоцетинкофакторной активности), Ø индуцированная ристоцетином агглютинация тромбоцитов Ø антигенная структура фактора Виллебранда, связанного с фактором VIII (VIII-ф. В).

Лабораторная диагностика БВ Ø Ø Ø Нормальное или субнормальное количество тромбоцитов; Нормальная или субнормальная коагуляция по тестам АЧТВ и ПТВ; Удлинение времени капиллярного кровотечения при нормальном или субнормальном времени свертывания; Удлинение времени ристомицин-индуцированной агрегации (РКА); Низкая адгезия Тр к стеклонитям или стеклянным бусинам; Снижение активности фактора Виллебранда (менее 50%) или действие ингибиторов.

Лечение БВ Ø Лечение зависит от типа заболевания. Выделяют два основных способа. При первом используются препараты плазмы с высоким содержанием фактора Виллебранда или препараты фактора VIII. Концентрат фактора VIII в комплексе с фактором фон Виллебранда(антигемофильный фактор, более известный как Humate-P). В легких случаях может быть достаточно одного введения, при тяжелых травмах и операциях препараты вводят дважды в сутки 2 -3 дня.

Лечение БВ Второй подход к лечению применим для легких форм. Пациентам назначается десмопрессин - 1 -desamino- 8 -D-аргинин вазопрессина, DDAVP(десмопрессин ацетат, Stimate), который повышает уровень фактора Виллебранда в плазме пациента, путем высвобождения ФВ, который содержится в тельцах Вэйбел-Паладе, находящихся в эндотелиальных клетках. Существует опасность привыкания при использовании десмопрессина более 2 -х суток.

Лечение БВ Неспецифическая терапия Ø • Дицинон Ø • Аминокапроновая кислота Ø • Местные кровоостанавливающие средства Ø • Механический гемостаз
")
Нарушения тромбоцитарного звена гемостаза (тромбоцитопении, тромбоцитопатии)

Тромбоцитопении количество тромбоцитов ниже 150 х109/л Ø практически значимо снижение числа тромбоцитов ниже 100 х109/л, а угроза развития серьезных геморрагий возникает при их уровне ниже 30 x 109/л (число Франка). Ø являются достаточно частыми причинами кровоточивости у детей (4, 5 -7, 5 на 100 000 населения), из них 47% приходится на идиопатическую тромбоцитопеническую пурпуру (ИТП). Ø

Тромбоцитопатии расстройства гемостаза, обусловленные качественной неполноценностью кровяных пластинок при нормальном их количестве. Ø Распространение даже первичных наследственных тромбоцитопатий не установлено, но несомненно, что это самая частая генетически обусловленная патология системы гемостаза. В большинстве случаев так называемой семейной кровоточивости неясного генеза можно подозревать наследственные тромбоцитопатии. Ø Частота наследственных тромбоцитопатий в популяции достигает 5% и более. Ø
;")
Тромбоцитопении Первичные 1. 2. Ø Ø Ø 3. Ø Ø 4. ИТП (болезнь Верльгофа); наследственные врожденный а (гипо-) - мегакариоцитоз (в сочетании с пороками развития (TAR-синдром: ТП + аплазия лучевой кости + дефицит факторов VII и X) или без них, тромбоцитопения при неэффективном тромбоцитопоэзе (ингибирование тромбопоэтина), тромбоцитолитическая ТП — синдромы Бернара-Сулье, Мея. Хеглина, «серых тромбоцитов» , Вискотта-Олдрича, Мерфи. изоиммунные врожденная — при несовместимости плода и матери по тромбоцитарным антигенам, посттрансфузионная — после переливаний крови и тромбоцитной массы; врожденная трансиммунная (транзиторная тромбоцитопения новорожденных от матерей, больных ИТП, системной красной волчанкой). Вторичные недостаточное образование в КМ (лейкозы, лучевая болезнь, гипоплазия костного мозга, болезни Гоше, Нимана— Пика, мукополисахаридозы), 2. укорочение продолжительности жизни или повышенная агрегация и потребление тромбоцитов под влиянием антитромбоцитарных антител (при вирусном гепатите В, СПИДе, при системных васкулитах, СКВ, в острый период инфекционных заболеваний - особенно часто при перинатальных вирусных инфекциях, болезнях, сопровождающихся спленомегалией и гиперспленизмом ) 3. повышенное потребления в тромбах и агрегатах клеток крови (ДВС-синдром, ГУС, болезнь Мошковица, врожденных аномалиях сосудов - гемангиомах). 1.

ИТП изолированное снижение числа Тр менее 150 х109/л, нормальное или повышенное количестве МКЦ в КМ, наличие на поверхности Тр и в сыворотке крови антитромбоцитарных АТ. Ø одинаковая частота этого заболевания у мальчиков и девочек, 1: 10 000 в год. Ø у детей любого возраста, но чаще у дошкольников и школьников. Ø сезонность в возникновении болезни: более частые случаи зимой и весной, несколько реже — осенью, и наиболее редко - летом. Ø

Этиология: Ø вирусная, реже - бактериальная инфекции, возникновение обычно через 2 -3 недели после начала острых респираторных заболеваний, краснухи, ветряной оспы, кори, реже гриппа и аденовирусной инфекции, инфекционного мононуклеоза Ø профилактические прививки ( АКДС, полиомиелитная, коревая вакцины) Ø введение гамма—глобулина Ø психические и физические травмы Ø прием лекарств (салицилатов, антибиотиков, сульфаниламидов, дигоксина, ПАСК, солей золота, гипотиазида) Ø особенности реактивности Ø наследственная предрасположенность Патогенез: Ø Повышенное разрушение нагруженнных аутоантителами тромбоцитов клетками СМФ Ø снижение продолжительности жизни тромбоцитов до нескольких часов и даже минут.

Этиология и патогенез ИТП Бактериальная или вирусная инфекция Профилактическ ие прививки Л С Переохлажден ия и инсоляции Операци и, травмы H. pylori Поступление АГ в организм Оседание АГ на тромбоцитах Иммунный ответ Ig G Реакция Аг-АТ на поверхности тромбоцитов Разрушение тромбоцитов в селезенке Тромбоцитопен ия Снижение ангиотрофической функции тромбоцитов Снижение концентрации серотонина в крови Невозможность ретракции кровяного сгуствка Петехиально-пятнистый (синячковый) тип кровоточивости

Клиника ИТП Выделяют «сухую» и «влажную» пурпуры. Кожный геморрагический синдром - у 100% больных. Характерные черты: 1) спонтанность возникновения, преимущественно по ночам, и неадекватность их степени внешнего воздействия (при травмах); 2) полиморфность (наряду с подкожными кровоизлияниями разной величины — экхимозами — имеются мелкоточечные — петехии); 3) полихромность(как правило, одновременно обнаруживаются на коже геморрагии разной окраски — от красновато-синеватых до зеленых и желтых); 4) несимметричность. «Излюбленной» локализации кожного геморрагического синдрома нет. Ø Кровотечения: носовые, желудочно-кишечные (мелена), кровотечения из десен, гематурия, кровотечения из лунки удаленного зуба и после других «малых» хирургических вмешательств. У девочек — мено- и метроррагии. Ø Кровоизлияния во внутренние органы: в сетчатку глаз, стекловидное тело, поджелудочную железу, яичники, внутреннее ухо и др. кровоизлияние в мозг(1— 3%). Ø Ø

Формы ИТП 1. Острая ф. – чаще встречается у детей 2 -8 лет, м: д=1: 1; геморрагический с-м возникает внезапно через 2 -3 нед. после вир. инфекции или иммунизации; Тр. нормализуются в течение 2 -8 нед. без спец. терапии. Ø 2. Хроническая ф(снижение Тр. более 6 мес. ) начинается постепенно без явного провоцирующего ф. , д>м, только в 10 -20% может начинаться как острая форма Ø

Факторы риска хронического течения ИТП Ø 1. длительность тромбоцитопении > 2 -4 нед. от момента диагностики Ø 2. Тр. > 50000/мкл Ø 3 женский пол Ø 4. возраст >10 лет

Периоды тромбоцитопенической пурпуры Геморрагический криз характеризуется выраженным синдромом кровоточивости, значительными изменениями лабораторных показателей. 2. Клиническая ремиссия. Исчезает геморрагический синдром, сокращается время кровотечения, уменьшаются вторичные изменения в свёртывающей системе крови, но тромбоцитопения сохраняется. 3. Клинико-гематологическая ремиссия. Отсутствие кровоточивости и нормализация лабораторных показателей. 1.

Аутоиммунная тромбоцитопения может быть атипичной презентацией СКВ, миелодиспластического синдрома, неспецифического язвенного колита и др.

Лабораторная диагностика ИТП Ø 1. общий ан. крови (изолированная тромбоцитопения без изменения эритроцитов, лейкоцитов и норм. ретикулоцитах) Ø 2. определение антигенспецифичных антитрооцитарных антител Ø 3. определение среднего объема Тр. Ø 4. пункция костного мозга Ø 5. ВИЧ-тест Ø 6. антинуклеарный фактор Ø 7. проба Кумбса

Лечение ИТП Ø I. острая форма – при миним. пурпуре или отсутствии геморраг. с-ма, отсутствии кровотечений со слизистых и уровне Тр. не < 30000/мкл – выжидательная тактика без применения специфической терапии. Симптоматическая терапия (эпсилон- аминокопроновая к-та, децинон, этамзилат, пр-ты Са)
 Ø II. Специфическая терапия. Ø 1. ВВИГ (курсовая доза) -1000 мг/кг")
Лечение ИТП (продолжение) Ø II. Специфическая терапия. Ø 1. ВВИГ (курсовая доза) -1000 мг/кг однократно или 2000 мг/кг в 2 -5 дней 2. кортикостероиды станд. доза – 1 -2 мг/кг/сутки 21 день - высокие дозы (парантерально) -4 -8 мг/кг/с 7 дн. - пульс-терапия – в/в метипред 10 -30 мг/кг 1 р. в день 3 -7 дней -
 Ø 3. Анти-Д-иммунноглобулин (из сыворотки людей, содержащей RH-АТ) – антиэритроцитарные Ig.")
Лечение ИТП (продолжение) Ø 3. Анти-Д-иммунноглобулин (из сыворотки людей, содержащей RH-АТ) – антиэритроцитарные Ig. G АТ средняя доза – 50 мкг/кг однократно или в 2 -5 дней (в/в или в/м) Ø 4. Спленэктомия (редко у детей до 5 лет из-за риска бактериальных инфекций)
 Ø 5. Цитостатики (циклофосфан, азатиоприн, колхицин, винкристин) Ø 6. Плазмоферез Ø")
Лечение ИТП (продолжение) Ø 5. Цитостатики (циклофосфан, азатиоприн, колхицин, винкристин) Ø 6. Плазмоферез Ø 7. Интерферон α 2 Ø 7. Даназол (синтетический андроген) Ø 8. Циклоспорин А.

Насилие над детьми Ø Частота случаев насилия над детьми в Швеции – 2%, Финляндии – 7, 7%, Германии – 10 -15% Ø США 1999 г. – 1100 случае смерти детей в результате насилия, из их 42% грудные дети

Данные, которые могут установить неслучайный характер травмы Ø Данные об обстоятельствах появление признаков неполные, нечеткие, противоречивые Ø Выраженные изменения, в которых обвиняется сам ребенок или другие дети Ø Запоздалое обращение к врачу Ø Сочетанные изменения Ø Рецидивы неясного генеза Ø Частая смена лечащего врача

Гематомы – самые частые симптомы насилия над детьми Признаки насилия: - множественные гематомы «подозрительных участках» - конфигурация гематом - петхиальные кровоизлияния Ø

Конфигурация гематом Ø Характерные следы укусов Ø Следы захвата или отпечатки пальцев Ø Иногда отпечатки кольца Ø Фигурные гематомы от пряжки ремня, палки

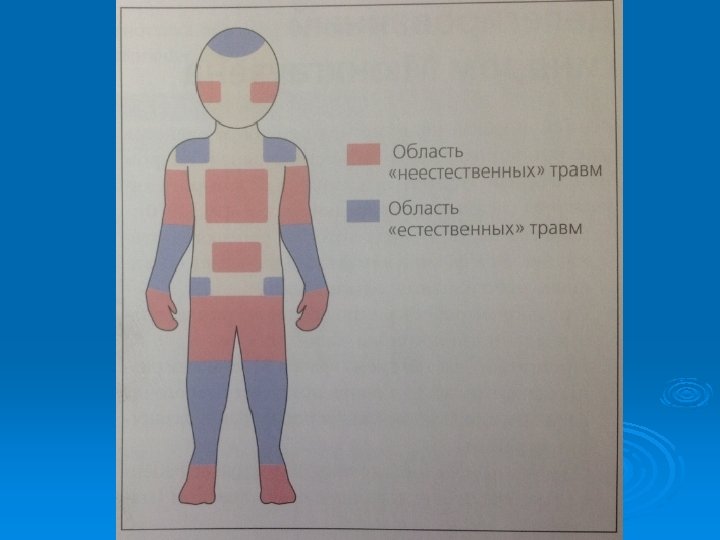
Распределение гематом Ø Неслучайный характер: грудная клетка, спина, ягодицы, гениталии, шеи, затылка, вентральная часть предплечий – защита от ударов, область лопаток, симметричные гематомы плечей и кистей Ø Не вызывает сомнений (если ребеное ходит): область лба, подбородка, носа, бедер, таза, внутренние поверхности лодоней, голени

Вазопатии Наследственные Собственно геморрагические ангиодисплазии (телеангиэктазия -болезнь Рандю-Ослера, телеангиэктатическая атаксия - синдром Луи-Бар и др. ). 2. Гемангиомы, протекающие с тромбоцитарными и коагуляционными нарушениями (синдром Казабаха-Мерритта, микроангиоматозы и др. ). 3. Формы с наследственной неполноценностью соединительной ткани, часто сочетающиеся с тромбоцитопатиями, дефицитом фактора Виллебранда и др. (синдромы Элерса-Данло, Марфана и др. ) - объединенные в группу геморрагические мезенхимальные дисплазии (Баркаган, 1988). 1. Приобретенные 1. 2. 3. 4. Геморрагический и другие виды аллергических васкулитов Симптоматические васкулиты при коллагенозах, медикаментозных и пищевых аллергозах Инфекционные и токсические вазопатии Гиповитаминозные вазопатии – дефициты витаминов С, Р и др.
bf3825b266c127165f4f06d3032e2493.ppt