Геморрагические диатезы, ВИА.ppt
- Количество слайдов: 139



ГЕМОРРАГИЧЕСКИЕ ДИАТЕЗЫ Лекция для стомфака Кафедра внутренних болезней и поликлинической терапии Ом. ГМА Свертывание крови: формирование сгустка.

Геморрагический диатез – это клинико-гематологический синдром, характеризующийся кровоточивостью. Объединяет геморрагические и тромбогеморрагические заболевания, а также геморрагические синдромы при инфекционно-септических, иммунных, сердечно-сосудистых, неопластических заболеваниях, акушерской патологии, болезнях новорожденных.

При геморрагическом диатезе отмечается выраженная тенденция к повторным кровоизлияниям или кровотечениям, наступающим как самопроизвольно, так и под влиянием незначительных травм. Это может быть как основным проявлением болезни (гемофилия), так и осложнением какого-либо заболевания (лейкозы, апластическая анемия, нефрит и др. )

Звенья системы гемостаза: ü тромбоцитарное ü коагуляционное, ü сосудистое.

Гемостаз • . n n n Вазоконстрикция Активация тромбоцитов Агрегация тромбоцитов Активация коагуляции Рыхлый сгусток Стабильный сгусток

Свертывание крови. Формирование сгустка.

ТИПЫ КРОВОТОЧИВОСТИ 1. Гематомный – болезненные напряженные кровоизлияния в мягкие ткани, в суставы. Типичен для гемофилии А и В. 2. Петехиально-пятнистый (синячковый, микроциркуляторный). Характерен для тромбоцитопений, тромбоцитопатий, некоторых нарушений свертываемости крови (гипо- и дисфибриногенемий, дефицит факторов X, II, VII).

Гематома у больного гемофилией

ГЕМАРТРОЗ

ГЕМАРТРОЗ

Тромбоцитопения • Петехиальные кровоизлияния на ногах • Кровоизлияние в конъюнктиву • Типичные изменения эритроцитов (включения хроматина) у больного после спленэктомии

Тромбоцитопения Петехии Пурпура

ТИПЫ КРОВОТОЧИВОСТИ 3. Смешанный синячково-гематомный – сочетание петехиально-пятнистой кровоточивости с появлением больших гематом (забрюшинных, в стенке кишечника и т. д. ) при отсутствии поражений суставов и костей либо единичными геморрагиями в суставы. При дефиците факторов протромбинового комплекса и фактора XIII, болезни Виллебранда, ДВС –синдроме, передозировке антикоагулянтов и тромболитиков.
,")
ТИПЫ КРОВОТОЧИВОСТИ 4. Васкулитно-пурпурный – геморрагии в виде сыпи или эритемы (на воспалительной основе), возможен нефрит, кишечные кровотечения; наблюдается при инфекционных и иммунных васкулитах, легко трансформируется в ДВС-синдром. 5. Ангиоматозный тип наблюдается при телеангиэктазиях, артериовенозных шунтах, ангиомах. Геморрагии определенной сосудистой локализации.
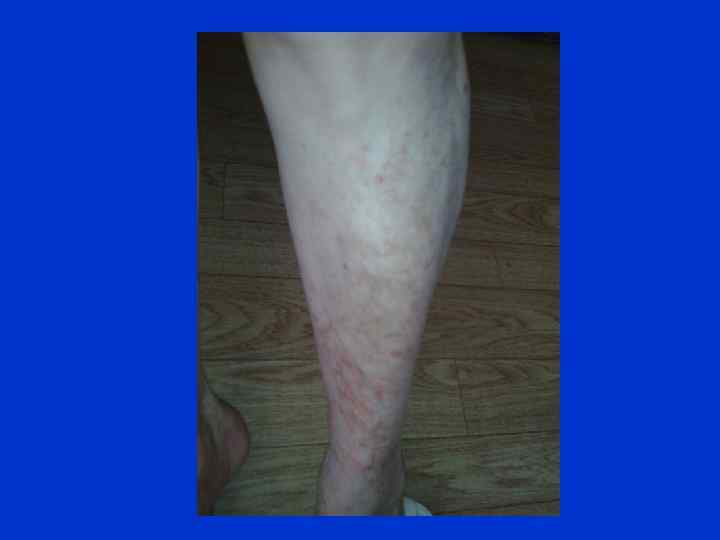

Васкулит

Васкулит

Экхимозы

Системный васкулит
")
КЛАССИФИКАЦИЯ ГЕМОРРАГИЧЕСКИХ ДИАТЕЗОВ (З. С. Баркаган, 1988)
. 1.")
1. Количественные и качественные изменения системы тромбоцитов: 1. 1. Болезнь Верльгофа (идиопатическая, иммунная). 1. 2. Симптоматические тромбоцитопении (инфекционно-токсические, медикаментозные, радиационные, гиперспленические, при лейкозах, аплазии при карциноматозе костного мозга). 1. 3. Тромбастения Гланцманна. 1. 4. Геморрагическая тромбоцитопения 1. 5. Тромбогемолитическая и тромбоцитопеническая пурпура (болезнь Мошковиц).
 – коагулопатии: 2. 1. Гемофилия А, В, С.")
2. Нарушения свертываемости крови (коагуляционный гемостаз) – коагулопатии: 2. 1. Гемофилия А, В, С. 2. 2. Гипопроакцелеринемия, гипопроконвертинемия (нарушение тромбинообразования). 2. 3. Гипопротромбинемия при механической желтухе, поражении печени; дикумариновая. 2. 4. Гипо-, афибриногенемия. 2. 5. Фибринолитическая пурпура.

3. Нарушение сосудистого звена системы гемостаза – вазопатии: 3. 1. Геморрагический васкулит (болезнь Шенлейна -Геноха). 3. 2. Геморрагическая пурпура (инфекционная, токсическая, нейровегетативная, трофическая). 3. 3. Дизовариальная пурпура (геморрагическая метропатия). 3. 4. С-авитаминоз (скорбут). 3. 5. Геморрагические телеангиэктазии (болезнь Рандю-Ослера, наследственный ангиоматоз). 3. 6. Ангиогемофилия (болезнь Виллебранда).

Из наследственных нарушений гемостаза 99% всех генетически обусловленных форм кровоточивости составляют 1. тромбоцитопатии, 2. гемофилия А, 3. болезнь Виллебранда, 4. гемофилия В, 5. телеангиэктазия.

Среди приобретенных форм преобладают : 1. тромбоцитопении вторичные, 2. тромбоцитопатии, 3. ДВС-синдром, 4. дефицит и ингибиция факторов протромбинового комплекса (патология печени, механическая желтуха, передозировка непрямых антикоагулянтов), 5. геморрагический васкулит. Все другие формы редки или очень редки.
; • Объем кровопотери;")
Особенности сбора анамнеза при геморрагическом синдроме • Вид кровотечения (местное, генерализованное); • Объем кровопотери; • Частота кровотечений; • Место проявления: кожа, слизистые, суставы и пр. ; • Спонтанное или после приема лекарств, травмы, хирургических вмешательств; • Семейный анамнез.

Физикальное обследование при геморрагическом синдроме Характеристика геморрагий: • петехии; • пурпуры; • экхимозы – «синяки» ; • гематомы; • гемартрозы; • гематурия • носовые кровотечения; • телеангиэктазии и ангиомы.

ПЕТЕХИИ точечные кровоизлияния в кожу или слизистые оболочки диаметром 1 -2 мм, обусловленные пропотеванием эритроцитов через стенку капилляров. Вначале имеют ярко-красную окраску, в последующем цвет изменяется до коричневатого. Петехии не возвышаются над поверхностью кожи и не пальпируются.
 - множественные кровоизлияния (петехии, экхимозы) в")
ПУРПУРА (лат. purpura, пурпурная ткань, багряница, порфира) - множественные кровоизлияния (петехии, экхимозы) в кожу и слизистые оболочки. - кожная сыпь, образующаяся после кровоизлияния в кожу и слизистые оболочки из капилляров.

пурпура Геморрагический васкулит
 Ребенок 5 лет после ОРВИ")
пурпура (геморрагический васкулит) Ребенок 5 лет после ОРВИ
")
множественные петехии на слизистой оболочке толстой кишки (колоноскопия)

кровоточивость, кровотечения

Обследование при подозрении на геморрагический диатез: • общий анализ крови • количественная и морфологическая оценка тромбоцитов в мазке периферической крови • АЧТВ – активированное частичное тромбопластиновое время характеризует целостность внутреннего и общего механизмов гемокоагуляции • ПВ – протромбиновое время выявляет функции внутреннего и общего механизмов свертывания крови • время кровотечения – для оценки целостности сосудов и функциональной активности тромбоцитов.

Дифференциально-диагностические признаки основных форм геморрагических диатезов 1. 2. 3. 4. 5. 6. 7. Тип кровоточивости Наследственность Число тромбоцитов Функция тромбоцитов Время кровотечения Время свертывания ДВС- синдром

ГЕМОФИЛИЯ 1. 2. 3. 4. 5. 6. 7. Тип кровоточивости - гематомный Наследственность - имеется Число тромбоцитов - норма Функция тромбоцитов - норма Время кровотечения - норма Время свертывания - удлинено ДВС- синдром - отсутствует

Коагулопатия Гематома, Кровоизлияние в сустав

БОЛЕЗНЬ ВИЛЛЕБРАНДА 1. 2. 3. 4. Тип кровоточивости - смешанный Наследственность - имеется Число тромбоцитов - норма Функция тромбоцитов – нарушение адгезии 5. Время кровотечения - удлинено 6. Время свертывания - удлинено 7. ДВС- синдром - отсутствует

ТРОМБОЦИТОПЕНИЯ 1. Тип кровоточивости – петехиальнопятнистый 2. Наследственность - отсутствует 3. Число тромбоцитов - снижено 4. Функция тромбоцитов – норма или нарушена 5. Время кровотечения - удлинено 6. Время свертывания - норма 7. ДВС- синдром - отсутствует

ТРОМБОЦИТОПАТИЯ • Тип кровоточивости – петехиальнопятнистый • Наследственность – часто имеется • Число тромбоцитов – норма или снижено • Функция тромбоцитов –нарушена • Время кровотечения - удлинено • Время свертывания - норма • ДВС- синдром - отсутствует

Тромбоцитопатическая пурпура

БОЛЕЗНЬ РАНДЮ-ОСЛЕРА 1. Тип кровоточивости ангиоматозный 2. Наследственность – имеется 3. Число тромбоцитов – норма 4. Функция тромбоцитов –норма 5. Время кровотечения - норма 6. Время свертывания - норма 7. ДВС- синдром – может быть –

Болезнь Рандю-Ослера

Наследственная геморрагическая телеангиэктазия
;")
Общие принципы лечения 1. Не показаны препараты, отрицательно влияющие на функцию тромбоцитов (например, аспирин); пункции глубоких вен, внутримышечные инъекции, длительное обездвиживание. 2. Профилактика травм (особенно черепно-мозговой). 3. После выявления конкретного дефекта – специфическая заместительная терапия. 4. При угрожающих жизни кровотечениях в качестве временной меры (до выявления конкретного фактора свертывания) возможно переливание свежезамороженной донорской плазмы (10 -20 мл/кг) или криопреципитата (5 -7 мл/кг) для восполнения недостаточности плазменных факторов свертывания.

Факторы свертывания крови
 Шенлейн-Геноха системный васкулит, поражающий сосуды микроциркуляторного русла (артериолы, капилляры и")
ГЕМОРРАГИЧЕСКИЙ ВАСКУЛИТ Болезнь (пурпура) Шенлейн-Геноха системный васкулит, поражающий сосуды микроциркуляторного русла (артериолы, капилляры и посткапиллярные венулы), с характерным отложением в их стенке иммунных депозитов, состоящих преимущественно из иммуноглобулинов А (Ig. A); клинически проявляется кожной геморрагической сыпью в сочетании с поражением суставов, желудочнокишечного тракта и почек.

ГЕМОРРАГИЧЕСКИЙ ВАСКУЛИТ: эпидемиология Заболеваемость составляет 13– 20 случаев на 100 000 человек. Развивается в возрасте от 5 месяцев до 89 лет, наиболее часто – у детей в возрасте 4– 6 лет, заболеваемость составляет 70 на 100 000 детей. С возрастом частота постепенно снижается. Мужчины и женщины болеют одинаково часто. Отмечена связь со временем года: наибольшая заболеваемость наблюдается осенью, зимой и весной, в половине случаев у детей предшествует острая респираторная инфекция.
,")
ГЕМОРРАГИЧЕСКИЙ ВАСКУЛИТ Этиология неизвестна. Возможна связь со стрептококковой и вирусной инфекцией (вирус гепатита В), микоплазмами, вакцинами, укусами насекомых, пищевыми и лекарственными аллергенами, охлаждением, сенсибилизацией. Связь заболевания с аллергической наследственностью и HLA В 35.
, кожных высыпаний")
ГЕМОРРАГИЧЕСКИЙ ВАСКУЛИТ Клиническая картина. Острое начало с общих симптомов (слабость, недомогание, лихорадка), кожных высыпаний (пурпуры) и суставного синдрома; в начале болезни часто – абдоминальный синдром. Частота симптомов: • • кожная геморрагическая сыпь — 100%; суставной синдром — 75%; абдоминальный синдром — 65%; поражение почек — 40%.
 размерами от 3 до")
ГЕМОРРАГИЧЕСКИЙ ВАСКУЛИТ Кожные высыпания – двусторонняя симметричная геморрагическая сыпь (пурпура) размерами от 3 до 10 мм, не бледнеет при надавливании (отличие от эритемы). Типичная локализация – голени и стопы, нередко – бедра, ягодицы, туловище, руки, редко – лицо. Постепенно бледнеют, трансформируются в коричневые пигментные пятна (вследствие гемосидероза) или исчезают. Склонность к рецидивированию после длительного пребывания в вертикальном положении.

ГЕМОРРАГИЧЕСКИЙ ВАСКУЛИТ Реже отмечаются экхимозы — крупные кожные геморрагии неправильной формы диаметром свыше 10 мм. Типичной локализацией экхимозов являются места, подвергающиеся механической компрессии (резинка носков, тугой ремень, манжетка тонометра). Этот феномен – аналог симптома Кончаловского–Румпеля–Лееде или симптома жгута.

ГЕМОРРАГИЧЕСКИЙ ВАСКУЛИТ 1. Тип кровоточивости – васкулитнопурпурный 2. Наследственность – отсутствует 3. Число тромбоцитов – норма 4. Функция тромбоцитов –норма 5. Время кровотечения - норма 6. Время свертывания - норма 7. ДВС- синдром - имеется

Геморрагический васкулит
 Ребенок 5 лет после ОРВИ")
пурпура (геморрагический васкулит) Ребенок 5 лет после ОРВИ

Болезнь Шенлейн-Геноха

Суставной синдром Второй по частоте и важности признак. Поражаются коленные и голеностопные суставы: быстрое возникновение болей, их летучесть, несоответствие выраженности болевых ощущений и скудности объективных признаков воспаления. Стойкой деформации и нарушения подвижности суставов не бывает.

Абдоминальный синдром – приступообразные боли в животе по типу кишечных колик, локализуются вокруг пупка, реже – в правой подвздошной или эпигастральной области, имитируя картину аппендицита или язвы желудка, панкреатита, острой кишечной непроходимости. На высоте боли возможны гематомезис, мелена. Хирургические осложнения: инвагинация, кишечная непроходимость, перфорация кишечника с развитием перитонита.

Поражение почек Гломерулонефрит. Ведущий симптом – гематурия в сочетании с умеренной протеинурией (менее 1 г/сут). У детей в дебюте может быть макрогематурия, не имеющая прогностического значения. У взрослых может быть нефротический синдром или быстропрогрессирующий нефрит.
. В детском возрасте может")
ГЕМОРРАГИЧЕСКИЙ ВАСКУЛИТ Течение заболевания может быть острым и хроническим (рецидивирующим). В детском возрасте может протекать тяжело в виде молниеносной пурпуры. Наряду с рецидивирующими формами существуют легкие, заканчивающиеся выздоровлением.

ГЕМОРРАГИЧЕСКИЙ ВАСКУЛИТ Тактика ведения Все больные в стадии обострения нуждаются в госпитализации, следует избегать охлаждения, длительного стояния и ходьбы. Необходимо исключить вероятные аллергические воздействия (лекарственные вещества, пищевые аллергены, введение сывороток и вакцин).

Лечение геморрагического васкулита Глюкокортикоиды в высоких дозах. При поражении кожи эффективны колхицин, сульфасалазин. При поражении почек – ГКС, цитостатики (циклофосфамид), плазмаферез.
 В 2005 г. опубликован отчет Всемирной федерации гемофилии (ВФГ)")
Нарушения свертываемости крови (коагуляционный гемостаз) В 2005 г. опубликован отчет Всемирной федерации гемофилии (ВФГ) о популяционном исследовании, включившем 98 стран на всех континентах и 88% всего населения Земли. Выявлено 131 264 больных гемофилией типов А и В, 45 001 человек с болезнью фон Виллебранда, и 16 735 – с другими нарушениями свертываемости крови. Всего в мире – более 193 тыс. больных с нарушениями свертываемости крови.

ГЕМОФИЛИЯ С 1989 г. день 17 апреля объявлен Всемирным днем гемофилии.

ГЕМОФИЛИЯ Гемофилия – это наследственное заболевание, проявляющееся недостаточностью факторов свертываемости крови VIII (гемофилия типа А) или IX (гемофилия типа В). Наследование данного признака происходит по Х-хромосоме.


ГЕМОФИЛИЯ
")
Королева Виктория (1819 -1901)
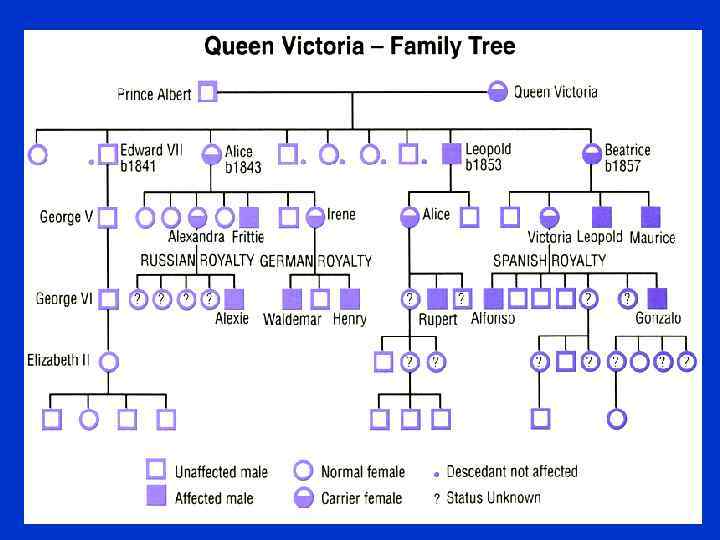

АЛЕКСЕЙ

Гемофилия А Ген гемофилии локализуется в Ххромосоме, в связи с чем женщины – кондукторы этого заболевания, имеющие вторую нормальную Х-хромосому, как правило, не страдают кровоточивостью, но активность фактора VIII у них снижена в среднем в 2 раза по сравнению с нормой.

Гемофилия А Количество фактора VIII крови измеряется в процентах. За основу берется среднее количество фактора в популяции, выраженное в «единицах на 1 мл» или « 100 ед. на децилитр (100 U/dl)» . Таким образом, 100% – не максимальное значение, а среднее. Уровень фактора VIII в норме составляет 50 – 180%.

Гемофилия А подразделяется на группы в зависимости от уровня фактора VIII: 1. от 0 до 1% – крайне тяжелая форма; 2. от 1 до 2% – тяжелая форма; 3. от 2 до 5% – форма средней тяжести; 4. выше 5% – легкая форма с возможностью тяжелых и даже смертельных кровотечений при травмах и хирургических вмешательствах. 25– 50% имеет 1/3 носителей аномального гена. 50– 180% средний уровень в популяции, более высокий встречается у беременных.
.")
Гемофилия В - наследственный геморрагический диатез, обусловленный дефицитом активности фактора IX (плазменного компонента тромбопластина). Наследуется по рецессивному, сцепленному с Х-хромосомой типу. Мутирует этот ген в 7 -10 раз реже, чем ген фактора VIII. Заболеваемость гемофилией типа В ниже в 3– 5 раз, чем типа А.
 свертывания крови. Имеет аутосомный характер наследования")
Гемофилия С связана с дефицитом фактора XI (РТАфактор) свертывания крови. Имеет аутосомный характер наследования и проявляется легкой или умеренной склонностью к кровотечениям, однако чаще протекает асимптомно.

КЛИНИКА ГЕМОФИЛИИ При гемофилии отмечается возрастная эволюция симптомов болезни. У большинства больных в первый год жизни геморрагических проявлений нет, в связи с чем заболевание распознается лишь на 2 -3 году жизни, а при легкой форме – во взрослом возрасте во время травмы или хирургического вмешательства.

КЛИНИКА ГЕМОФИЛИИ Главным и единственным признаком является склонность к кровотечениям. • Тяжелая форма – часты спонтанные кровотечения. • Среднетяжелая форма – возможны спонтанные кровотечения после незначительных травм. • Легкая форма – склонность к кровоточивости после травм и операций, спонтанных кровотечений не бывает. При легкой форме заболевания диагноз часто ставится впервые в уже зрелом возрасте.
, часто")
КЛИНИКА ГЕМОФИЛИИ Наиболее часто спонтанные и травматические кровоизлияния проявляются гемартрозами (70– 80% случаев), часто образуются подкожные гематомы (10– 20%). Гемартрозы – продолжительность острого периода и последующую деформацию сустава можно свести к минимуму, если быстро начать заместительную терапию и на 2 -3 суток иммобилизировать сустав.
, локтевых (30%), голеностопных (10%) суставах. Плечевые и тазобедренные")
ГЕМАРТРОЗ – в коленных (45% случаев), локтевых (30%), голеностопных (10%) суставах. Плечевые и тазобедренные суставы поражаются значительно реже (в 2– 3% случаев).

КЛИНИКА ГЕМОФИЛИИ Любая травматизация у больного гемофилией может вызвать, хотя и неинтенсивное, но непрекращающееся кровотечение. Запрещаются внутримышечные инъекции. Иммунизацию и введение лекарств нужно производить подкожно, тонкой иглой, обеспечив 5 -минутное давление на место инъекции.

КЛИНИКА ГЕМОФИЛИИ Возможны частые носовые кровотечения, кровотечения из ротовой полости (например, прикусывании языка, травме десен). Возможна упорная гематурия и наличие крови в кале. Выделение крови даже низкой интенсивности, но продолжительное и стойкое, приводит к анемизации больных. Наиболее опасны для жизни внутренние кровотечения (в забрюшинную клетчатку) и кровоизлияния в полость черепа.

КЛИНИКА ГЕМОФИЛИИ Кровотечения при гемофилии прекращаются после заместительной терапии соответствующим фактором свертывания крови, причем ее следует начинать как можно раньше, чтобы свести к минимуму осложнения. Больным гемофилией противопоказаны аспирин и нестероидные противовоспалительные средства.

КЛИНИКА ГЕМОФИЛИИ Клиническая картина гемофилии в остальном связана с частой рецидивирующей кровопотерей и обусловлена анемизацией больных. Как и для популяции в целом, главными причинами смерти пациентов с гемофилией являются онкологические и сердечно-сосудистые заболевания. Однако у больных гемофилией очень часто причинами смерти становятся травматические кровоизлияния в полость черепа, а также ВИЧ-инфекция и поражение печени вирусом гепатита С, связанные с необходимостью частых гемотрансфузий.

КЛИНИКА ГЕМОФИЛИИ Среди осложнений гемофилии следует назвать различные проявления суставного геморрагического синдрома (контрактуры, переломы, артропатию), кровоизлияния в мягкие ткани (опухолевидные образования, инфицирование), заражение трансмиссивными агентами (вирусы гепатитов А, В, С, ВИЧ, парвовирус В 19 и др. ), а также формирование ингибиторов (антител) к факторам свертывания с резким снижением эффективности лечения.
, •")
Лабораторная диагностика • Увеличение АЧТВ (активированное частичное тромбопластиновое время, норма 25 -36 с), • нормальное ПВ (протромбиновое время, норма 11– 14 с) • нормальное время кровотечения (по Айви 2, 5– 9, 5 мин).

Лабораторная диагностика Факторы VIII и IX участвуют только во внутренней активации гемостаза. Факторы VIII (антигемофильный глобулин) и IX (Кристмаса) в комплексе с ионами кальция и фосфолипидами создают условия для активации X фактора и формирования протромбиназного комплекса.

Лабораторная диагностика Данный факт обусловливает такую особенность коагулограммы при гемофилии, как нормальные протромбиновый показатель и тромбиновое время при удлиненном времени свертывания цельной крови, кальцифицированной цитратной плазмы и активированного частичного тромбопластинового времени (АЧТВ).

Диагностика • Наиболее достоверным в диагностике как заболевания, так и носительства гемофилии является молекулярно-генетическое исследование. Методика выявления полиморфизма ДНК позволяет диагностировать нарушения генов факторов свертывания с достоверностью >99%. Ее можно использовать в пренатальной диагностике (исследуя ДНК, экстрагированную из клеток ворсин хориона на 10 -й неделе беременности или позднее). • Методика ПЦР (полимеразной цепной реакции), позволяющая выявлять конкретные изменения в хромосомах: делецию, инверсию и т. п. • Составление подробного генеалогического древа.

ЛЕЧЕНИЕ Лечение гемофилии складывается из купирования геморрагического синдрома и лечения его осложнений, а также сопутствующих состояний, осложняемых основным заболеванием. Гемофилия представляется отличным кандидатом для молекулярно-генетического вмешательства.

ЛЕЧЕНИЕ Субстанции, содержащие фактор свертываемости, по возрастанию концентрации можно расположить в следующем порядке: цельная кровь, плазма, криопреципитат, препарат фактора. Наиболее богаты фактором VIII рекомбинантные препараты "Kogenate FS", "Helixate FS", "Recombinate r. AHF" американского производства, швейцарский "Advate r. AHF PFM", содержащие более 4000 ЕД/мг действующего вещества, а также препарат "Re. Facto" шведского производства с концентрацией 13000 ЕД/мг. Они свободны от человеческих или животных материалов, а потому безопасны с эпидемиологической точки зрения.

ЛЕЧЕНИЕ Введение препарата фактора свертываемости должно осуществляться как можно быстрее после начала кровотечения или только при подозрении на него, так как эффективность терапии и скорость восстановления прямо пропорциональны времени промедления. Идеально ввести препарат фактора в течение первых 2 часов. Нельзя применять лекарственные средства, нарушающие функцию тромбоцитов (в первую очередь НПВП). Внутривенные инъекции должны быть максимально аккуратными, производиться только при необходимости. Внутримышечные инъекции запрещены.

ЛЕЧЕНИЕ Главным осложнением терапии препаратами, повышающими свертываемость крови, считаются инфекционные осложнения: передача реципиенту ВИЧ, вирусов гепатитов А, В, С, парвовируса В 19, прионового агента болезни Крейцфельдта-Якоба. Прионовая болезнь Крейтцфельдта–Якоба (Creutzfeldt–Jakob), сходная с «коровьим бешенством» , вызывает спонгиозную трансформацию головного мозга людей. Ее инкубационный период составляет многие годы, а инфекционный агент представляет собой видоизмененный естественный представитель клеток человека – шаперон, в связи с чем его практически невозможно обнаружить.

ЛЕЧЕНИЕ Часто в нетяжелых случаях гемофилии А и почти во всех случаях болезни фон Виллебранда вместо препарата фактора свертываемости крови применяют десмопрессин – синтетическое производное антидиуретического гормона. Другими средствами уменьшения кровоточивости у пациентов с гемофилией являются транексамовая и аминокапроновая кислоты.

ПРОГНОЗ Хотя в современном мире еще не существует способа радикального излечения от гемофилии, заболевания, считавшегося ранее смертельным, наличие множества методов поддержания стабильного состояния организма и лечения осложнений этого заболевания позволяет больному человеку не выпадать из жизни общества, принимать в ней активное и полноценное участие.
: 1. 1. Болезнь Верльгофа (идиопатическая,")
Количественные и качественные изменения системы тромбоцитов (тромбоцитопении и тромбоцитопатии): 1. 1. Болезнь Верльгофа (идиопатическая, иммунная). 1. 2. Симптоматические тромбоцитопении (инфекционно-токсические, медикаментозные, радиационные, гиперспленические, при лейкозах, аплазии при карциноматозе костного мозга). 1. 3. Тромбастения Гланцманна. 1. 4. Геморрагическая тромбоцитопения 1. 5. Тромбогемолитическая и тромбоцитопеническая пурпура (болезнь Мошковиц).

ТРОМБОЦИТОПЕНИИ заболевания, при которых количество тромбоцитов ниже нормы – 150· 109/л. Если количество тромбоцитов превышает 50· 109/л, геморрагический диатез наблюдается редко. Причины: 1. Повышенное разрушение тромбоцитов. 2. Повышенное потребление тромбоцитов. 3. Недостаточное образование тромбоцитов.

ТРОМБОЦИТОПЕНИИ 1. Наследственные тромбоцитопении. При многих наследственных тромбоцитопениях наблюдается изменение различных функциональных свойств тромбоцитов, что дает основание относить эти болезни к группе тромбоцитопатий. Редко являются истинными, связанными с нарушением активности ферментов гликолиза или цикла Кребса, или нарушением образования тромбопоэтинов.

ТРОМБОЦИТОПЕНИИ 2. Приобретенные тромбоцитопении: ü иммунные; ü обусловленные механической травмой тромбоцитов (гемангиомы, спленомегалия и др. ); ü угнетение пролиферации клеток костного мозга при химических и радиационных повреждениях, апластических анемиях; ü замещение костного мозга опухолевой тканью; ü соматическая мутация (болезнь Маркиафавы. Микели); ü повышенным потреблением тромбоцитов (ДВСсиндром, тромбозы); ü недостаток витамина В 12 и фолиевой кислоты.

ИММУННЫЕ ТРОМБОЦИТОПЕНИИ Можно разделить на 4 группы: * Аутоиммунные * Гетероиммунные * Трансиммунные и изоиммунные * Аллоиммунные

ИММУННЫЕ ТРОМБОЦИТОПЕНИИ встречаются наиболее часто, причем у детей чаще гетероиммунные, у взрослых – аутоиммунные. В зависимости от антител делятся: 1) с антителами против антигенов тромбоцитов; 2) против антигенов мегакариоцитов; 3) против антигенов общего предшественника тромбоцитов, лейкоцитов и эритроцитов.
. Количество")
АУТОИММУННЫЕ ТРОМБОЦИТОПЕНИИ Продолжительность жизни клеток укорачивается до нескольких часов (вместо 7– 10 дней). Количество тромбоцитов, образующихся в единицу времени, значительно увеличивается – от 2 до 6 раз, также и количество мегакариоцитов. В основе патологического процесса лежит срыв иммунологической толерантности к собственному антигену, генетический дефект функции Т-супрессоров. Морфометрия тромбоцитов: большие размеры, малозернистые, «голубые» , пойкилоцитоз.

АУТОИММУННЫЕ ТРОМБОЦИТОПЕНИИ Содержание эритроцитов и Нв может быть нормальным или наблюдается постгеморрагическая анемия. Количество лейкоцитов либо нормальное, либо повышенное. Часто отмечается эозинофилия. Если отмечается ПАНЦИТОПЕНИЯ (тромбо-, лейкоцитопения и анемия), то антитела образуются к общему предшественнику трех ростков.
")
ГЕТЕРОИММУННЫЕ ТРОМБОЦИТОПЕНИИ Антитела вырабатываются против чужого антигена, фиксированного на поверхности тромбоцитов, (лекарства или вируса) или изменяется антигенная структура тромбоцитов, например, под влиянием вирусного воздействия.

ИЗОИММУННЫЕ ТРОМБОЦИТОПЕНИИ Наблюдаются у новорожденных в связи с несовместимостью по тромбоцитарным антигенам между матерью и ребенком, в отличие от гемолитической анемии может развиться как после первой, так и после второй беременности.

ТРАНСИММУННЫЕ ТРОМБОЦИТОПЕНИИ Аутоантитела матери, страдающей аутоиммунной тромбоцитопенией, проникают через плаценту и вызывают тромбоцитопению у ребенка.

АЛЛОИММУННЫЕ ТРОМБОЦИТОПЕНИИ Разрушение тромбоцитов связано с несовместимостью по одной из групповых систем крови либо в связи с транфузией реципиенту чужих тромбоцитов при наличии к ним антител; либо в связи с проникновением антител к ребенку матери, предварительно иммунизированной антигеном, отсутствующим у нее, но имеющимся у ребенка.

ТРОМБОЦИТОПЕНИИ Болезнь Верльгофа
 — хроническое волнообразно протекающее заболевание, представляющее собой")
Болезнь Верльгофа (хроническая иммунопатологическая тромбоцитопеническая пурпура) — хроническое волнообразно протекающее заболевание, представляющее собой первичный геморрагический диатез, обусловленный количественной и качественной надостаточностью тромбоцитарного звена гемостаза. В связи с многообразием тромбоцитопенических синдромов, осложняющих различные заболевания, термин утрачивает свое значение.

Болезнь Верльгофа Характеризуется элиминативной тромбоцитопенией, наличием гигантских тромбоцитов в кровотоке, мегакариоцитозом в костном мозге и обязательным присутствием антитромбоцитарных аутоантител. Симптомы описаны еще Гиппократом. Болезнь названа по имени немецкого врача П. Верльгофа, описавшего её в 1735 году.
 является причиной геморрагического синдрома")
Болезнь Верльгофа Заболевание наиболее часто (в 40 % случаев) является причиной геморрагического синдрома в гематологической практике. Распространенность болезни Верльгофа колеблется от 1 до 13 % на 100 000 человек. Большинство больных — женщины молодого и среднего возраста.
 и хронические")
Болезнь Верльгофа Классификация По течению выделяют острые (продолжающиеся менее 6 месяцев) и хронические формы ИТП. Последние подразделяются на варианты: с редкими рецидивами; с частыми рецидивами; непрерывно рецидивирующее течение. По периоду болезни выделяют обострение (криз), клиническую ремиссию (отсутствие каких-либо проявлений геморрагического синдрома при сохраняющейся тромбоцитопении) и клиникогематологическую ремиссию.
 типу")
Болезнь Верльгофа Типично внезапное появление геморрагического синдрома по микроциркуляторному (петехиально – пятнистому) типу у ребенка, который в других отношениях абсолютно здоров. Геморрагический синдром обычно представлен кожными геморрагиями (петехии, пурпура, экхимозы), кровоизлияниями в слизистые оболочки, кровотечениями из слизистых (носовые, десневые, из лунки удаленного зуба, маточные, реже —мелена, гематурия).

Болезнь Верльгофа Провоцирующие факторы, как правило, следующие: ОРВИ, детские инфекции (ветряная оспа, корь, краснуха), вакцинация, персистенция вирусов CMV, EBV, парвовирус В 19. При физикальном обследовании ребенка, кроме геморрагического синдрома, другие синдромы поражения (интоксикация, лимфоаденопатия, гепатоспленомегалия) не выявляются.

Болезнь Верльгофа Провоцирующие факторы, как правило, следующие: ОРВИ, детские инфекции (ветряная оспа, корь, краснуха), вакцинация, персистенция вирусов CMV, EBV, парвовирус В 19. При физикальном обследовании ребенка, кроме геморрагического синдрома, другие синдромы поражения (интоксикация, лимфоаденопатия, гепатоспленомегалия) не выявляются.

Болезнь Верльгофа Лечение. * Глюкокортикоиды из расчета 1 мг/кг с последующим снижением дозы и постепенной отменой препарата после нормализации количества тромбоцитов. * При неэффективности в течение 4 -5 мес показана спленэктомия. * При неэффективности – цитостатики. * Гемостатическая терапия.

ТРОМБОЦИТОПАТИИ общий термин для обозначения всех нарушений гемостаза, обусловленных качественной неполноценностью или дисфункцией кровянных пластинок. Подразделяются на: 1. Наследственные. 2. Приобретенные (симптоматические).
 • НАСЛЕДСТВЕННЫЕ И ВРОЖДЕННЫЕ ФОРМЫ")
КЛАССИФИКАЦИЯ ТРОМБОЦИТОПАТИЙ И ДИСФУНКЦИЙ ТРОМБОЦИТОВ (З. С. Баркаган, 1988) • НАСЛЕДСТВЕННЫЕ И ВРОЖДЕННЫЕ ФОРМЫ • ПРИОБРЕТЕННЫЕ

НАСЛЕДСТВЕННЫЕ И ВРОЖДЕННЫЕ ФОРМЫ I. ОСНОВНЫЕ ПАТОГЕНЕТИЧЕСКИЕ ГРУППЫ 1. Связанные с мембранными аномалиями (тромбастения Гланцманна, эссенциальная атромбия).
 болезни недостаточного пула хранения: - дефицит")
НАСЛЕДСТВЕННЫЕ И ВРОЖДЕННЫЕ ФОРМЫ 2. Внутриклеточные аномалии: а) болезни недостаточного пула хранения: - дефицит плотных (безбелковых) гранул (ТАРсиндром) - дефицит альфа-гранул (белковых) – синдром серых тромбоцитов. б) нарушение реакции высвобождения гранул и их компонентов: - дефицит циклооксигеназы; - дефицит тромбоксан-синтетазы; - другие патогенетические формы.
. 4. Дисфункции плазменного генеза:")
НАСЛЕДСТВЕННЫЕ И ВРОЖДЕННЫЕ ФОРМЫ 3. Смешанные тромбоцитарные нарушения (аномалия Вискотта-Олдрич). 4. Дисфункции плазменного генеза: - дефицит и аномалии фактора Виллебранда, - афибриногенемия, - другие плазменные нарушения. 5. Нарушения взаимодействия с коллагеном и субэндотелием: а) плазменного генеза – болезнь Виллебранда, б) аномалии коллагена – болезнь Элерса-Данло и другие мезенхимальные дисплазии.

НАСЛЕДСТВЕННЫЕ И ВРОЖДЕННЫЕ ФОРМЫ II. ФУНКЦИОНАЛЬНОМОРФОЛОГИЧЕСКИЕ ФОРМЫ 1. Формы с преимущественным нарушением агрегационной функции (дизагрегация) с сохранением реакции высвобождения: а) с развернутым нарушением агрегационной функции б) парциальные дизагрегационные тромбоцитопатии

НАСЛЕДСТВЕННЫЕ И ВРОЖДЕННЫЕ ФОРМЫ 2. Формы с нарушением реакции высвобождения и отсутствием второй волны агрегации - аспириноподобный синдром. 3. Болезни недостаточного пула хранения (дефицит гранул и их компонентов) с отсутствием второй волны агрегации: а) с недостатком плотных телец 1 типа и их компонентов – АДФ, серотонина, адреналина; б) с недостатком плотных телец 2 типа (альфагранул) и их компонентов – фактора 4 и его носителя, бета-тромбоглобулина, ростового фактора

НАСЛЕДСТВЕННЫЕ И ВРОЖДЕННЫЕ ФОРМЫ 4. С нарушением адгезии тромбоцитов к коллагену и стеклу (без закономерного нарушения физиологических видов агрегации). 5. С дефицитом и снижением доступности фактора 3 (без существенного нарушения адгезивной функции). 6. Сложные аномалии и дисфункции Тр. , сочетающиеся с другими генетическими дефектами. 7. Недостаточно идентифицированные формы.

Очень часто тромбоцитопатия сочетается с тромбоцитопенией и трудно решить, что в этих случаях является ведущим. Руководствуются следующими положениями: 1) к патиям относят те формы, при которых выявляются стабильные функциональные, морфологические и биохимические нарушения тромбоцитов, не исчезающие при нормализации количественных показателей.
 для патии характерно несоответствие между выраженностью геморрагического синдрома и степенью тромбоцитопении. 3) генетически")
2) для патии характерно несоответствие между выраженностью геморрагического синдрома и степенью тромбоцитопении. 3) генетически обусловленные формы патологии в подавляющем большинстве случаев относятся к патиям, особенно если они сочетаются с другими наследственными дефектами.
 если у той или иной категории больных качественный дефект тромбоцитов непостоянен и ослабляется")
4) если у той или иной категории больных качественный дефект тромбоцитов непостоянен и ослабляется или полностью исчезает после ликвидации тромбоцитопении, такую тромбоцитопатию следует считать вторичной. 5) все дисфункции тромбоцитов, выявляющиеся при иммунных тромбоцитопениях, рассматриваются как вторичные нарушения.

ТРОМБАСТЕНИЯ ГЛАНЦМАННА Семейно-наследственное заболевание. Болеют с детского возраста. Кровоточивость обусловлена отсутствием гликопротеида в оболочке тромбоцитов и значительным снижением содержания коллаген-глицеральдегидрофосфатдегидрогеназы, необходимой для взаимодействия этих клеток со стимуляторами агрегации и фибриногеном. Такие тромбоциты приклеиваются к волокнам коллагена, но не происходит дальнейшая агрегация.
 ТРОМБОЦИТОПАТИИ Отличаются сложностью генеза и большим разнообразием функциональных нарушений. При одних и")
ПРИОБРЕТЕННЫЕ (СИМПТОМАТИЧЕСКИЕ) ТРОМБОЦИТОПАТИИ Отличаются сложностью генеза и большим разнообразием функциональных нарушений. При одних и тех же заболеваниях и даже у одних и тех же больных в разные периоды болезни часто наблюдается мозаичность лабораторных признаков, неоднотипные сдвиги адгезивно-агрегационных, оагуляционных к и ретикулярных свойств кровянных пластинок.
 ТРОМБОЦИТОПАТИИ Исключение составляют лишь некоторые лекарственные и токсические формы, которые, подобно наследственным")
ПРИОБРЕТЕННЫЕ (СИМПТОМАТИЧЕСКИЕ) ТРОМБОЦИТОПАТИИ Исключение составляют лишь некоторые лекарственные и токсические формы, которые, подобно наследственным аномалиям, имеют четкую и стабильную функциональную маркировку. Так при В 12 -дефицитной анемии отмечается не только гипогенеративная тромбоцитопения, но и качественные изменения тромбоцитов, а именно, нарушение второй фазы агрегации (при воздействии коллагеном, АДФ и адреналином).

ПРИОБРЕТЕННЫЕ ТРОМБОЦИТОПАТИИ 1. При гемобластозах – дезагрегационные гиперрегенераторные: • формы потребления; • смешанные. 2. При В 12 -дефицитной анемии. 3. При уремии. 4. При ДВС-синдроме и активации фибробластоза.

ПРИОБРЕТЕННЫЕ ТРОМБОЦИТОПАТИИ 5. При циррозах, опухолях и паразитарных заболеваниях печени. 6. При макро- и парапротеинемиях. 7. При С-авитаминозах. 8. При гормональных нарушениях. 9. Лекарственные и токсигенные. 10. При лучевой болезни. 11. При массивных гемотрансфузиях, инфузиях реополиглюкина. 12. Больших тромбозах и гигантских ангиомах.

БОЛЕЗНЬ ВИЛЛЕБРАНДА установлено, что это не одно заболевание, а группа родственных по патогенезу геморрагических диатезов, обусловленных либо нарушением синтеза, либо качественными аномалиями, либо неправильным распределением аутосомных компонентов фактора VIII: ФВ и связанного с ним антигена – VIII: РАг.
 резко нарушен синтез этого фактора")
БОЛЕЗНЬ ВИЛЛЕБРАНДА При болезни Виллебранда I типа (классическая форма) резко нарушен синтез этого фактора и связанного с ним антигена в эндотелии сосудистой стенки, снижено содержание всех компонентов фактора VIII в плазме и тромбоцитах.

БОЛЕЗНЬ ВИЛЛЕБРАНДА Другие разновидности болезни Виллебранда имеют общую закономерность: функциональная активность фактора Виллебранда зависит от полимерной структуры комплекса фактора VIII. Наиболее высока эта активность при триплетном и квадратном строении данного надмолекулярного комплекса.

БОЛЕЗНЬ ВИЛЛЕБРАНДА По мере же уменьшения или, наоборот, чрезмерного увеличения размеров комплекса активность фактора Виллебранда снижается. При этом поразному нарушается взаимодействие фактора Виллебранда с тромбоцитами и ристомицином, а также взаимодействие тромбоцитов с сосудистой стенкой, что создает при всех вариантах этой патологии весьма пеструю картину гемостатических и функциональных нарушений.

БОЛЕЗНЬ ВИЛЛЕБРАНДА Это разнообразие болезни Виллебранда определяется, с одной стороны, сложностью надмолекулярной структуры комплекса VIII фактора и изменчивостью взаимодействия входящих в его состав разных активностей, а с другой – участием субъединиц фактора VIII во всех звеньях системы гемостаза (сосудистом, тромбоцитарном и коагуляционном) и неоднородностью нарушений в каждом из этих звеньев.
 нарушение синтеза компонентов фактора VIII или его")
БОЛЕЗНЬ ВИЛЛЕБРАНДА Ключевые параметры в патогенезе 1) нарушение синтеза компонентов фактора VIII или его высвобождения из сосудистого эндотелия, 2) нарушение мультимерной структуры ФВ, 3) нарушение взаимодействия фактора Виллебранда с тромбоцитами и распределение его между плазмой и тромбоцитами.
 сцепленность нарушений разных гемостатических функций, тогда как")
БОЛЕЗНЬ ВИЛЛЕБРАНДА Ключевые параметры в патогенезе 4) сцепленность нарушений разных гемостатических функций, тогда как при гемофилии А нарушается лишь изолированно коагуляционная функция фактора VIII, 5) нередкое сочетание с патологией микрососудов и другими дисмезенхимами, 6) аутосомное наследование болезни.

БОЛЕЗНЬ ВИЛЛЕБРАНДА В генетическом отношении болезнь Виллебранда также не однородна: наряду с частым аутосомнодоминантным наследованием описывают и аутосомно-рецессивные формы, протекающие у гетерозигот скрыто или бессимптомно, а у гомозигот – очень тяжело.
Геморрагические диатезы, ВИА.ppt