

Gemorragicheskie_diatezy.ppt663649808.ppt
- Количество слайдов: 28
 Геморрагические диатезы Классификация. Механизм кровоточивости при различных геморрагических диатезах. Клиника, диагностика, лечение, профилактика.
Геморрагические диатезы Классификация. Механизм кровоточивости при различных геморрагических диатезах. Клиника, диагностика, лечение, профилактика.
 Система гемостаза l Биологическая система, которая обеспечивает, с одной стороны – сохранение жидкого состояния крови, а с другой предупреждение и остановку кровотечений путем поддержки структурной целостности стенок кровеносных сосудов и достаточно быстрого тромбирования при повреждениях.
Система гемостаза l Биологическая система, которая обеспечивает, с одной стороны – сохранение жидкого состояния крови, а с другой предупреждение и остановку кровотечений путем поддержки структурной целостности стенок кровеносных сосудов и достаточно быстрого тромбирования при повреждениях.
 Диатез l Патологическая реактивность, характеризующаяся ненормальными реакциями на обычные раздражители и предрасположенностью к определенным заболеваниям
Диатез l Патологическая реактивность, характеризующаяся ненормальными реакциями на обычные раздражители и предрасположенностью к определенным заболеваниям
 Гемостаз реализуется, в основном, 3 -мя функциональноструктурными компонентами: l l l стенками кровеносных сосудов; клетками крови; плазменными ферментными системами – свертывающей, фибринолитической (плазминовой), калликреин кининовой системой и др.
Гемостаз реализуется, в основном, 3 -мя функциональноструктурными компонентами: l l l стенками кровеносных сосудов; клетками крови; плазменными ферментными системами – свертывающей, фибринолитической (плазминовой), калликреин кининовой системой и др.
 Факторы, участвующие в процессе свертывания крови: l l l l Фактор I фибриноген Фактор II протромбин Фактор III тканевой тромбопластин Фактор IV ионизирующий кальций Фактор V проакцелерин Фактор VI акцелерин Фактор VII прокорвентин Фактор VIII антигемофильный глобулин А Фактор IX антигемофильный глобулин В Фактор X протромбиназа Фактор XI антигемофильный глобулин С Фактор XII контактный Фактор XIII фибринстабилизирующий
Факторы, участвующие в процессе свертывания крови: l l l l Фактор I фибриноген Фактор II протромбин Фактор III тканевой тромбопластин Фактор IV ионизирующий кальций Фактор V проакцелерин Фактор VI акцелерин Фактор VII прокорвентин Фактор VIII антигемофильный глобулин А Фактор IX антигемофильный глобулин В Фактор X протромбиназа Фактор XI антигемофильный глобулин С Фактор XII контактный Фактор XIII фибринстабилизирующий
 Геморрагические диатезы l Группа заболеваний и синдромов, характеризующихся повышенной кровоточивостью в результате недостаточности одного или нескольких элементов гемостаза, склонность к повторным геморрагиям и кровоизлияниям, самопроизвольным или после незначительных травм
Геморрагические диатезы l Группа заболеваний и синдромов, характеризующихся повышенной кровоточивостью в результате недостаточности одного или нескольких элементов гемостаза, склонность к повторным геморрагиям и кровоизлияниям, самопроизвольным или после незначительных травм
 Геморрагические диатезы Первичные Дефект гемостаза – это как бы самостоятельное заболевание Вторичные Симптоматические, развивающиеся на фоне другого заболевания
Геморрагические диатезы Первичные Дефект гемостаза – это как бы самостоятельное заболевание Вторичные Симптоматические, развивающиеся на фоне другого заболевания
 Классификация ГЕМОРРАГИЧЕСКИХ ДИАТЕЗОВ: l ГД, обусловленные изменением стенок сосудов: наследственные приобретенные. l ГД, обусловленные изменением тромбоцитов: а) тромбоцитопатии; б)тромбоцитопении; наследственные; при обретенные. l ГД, обусловленные дефектами факторов свертывания крови: наследственные; приобретенные. l ГД, обусловленные избыточной активностью фибринолиза: а) наследственный дефицит а 2 антиплазмина; б) избыточное образование активаторов плазминогена; в) недостаточность инактивации активаторов плазминогена l ГД, обусловленные изменением нескольких факторов системы гемостаза: Болезнь Виллебранда ДВС синдром
Классификация ГЕМОРРАГИЧЕСКИХ ДИАТЕЗОВ: l ГД, обусловленные изменением стенок сосудов: наследственные приобретенные. l ГД, обусловленные изменением тромбоцитов: а) тромбоцитопатии; б)тромбоцитопении; наследственные; при обретенные. l ГД, обусловленные дефектами факторов свертывания крови: наследственные; приобретенные. l ГД, обусловленные избыточной активностью фибринолиза: а) наследственный дефицит а 2 антиплазмина; б) избыточное образование активаторов плазминогена; в) недостаточность инактивации активаторов плазминогена l ГД, обусловленные изменением нескольких факторов системы гемостаза: Болезнь Виллебранда ДВС синдром
 Типы кровоточивости: l l l I. ГЕМАТОМНЫЙ (массивные, глубокие, напряженные и очень болезненные кровоизлияния в крупные суставы, мышцы, подкожную и забрюшинную клетчатку, серозные оболочки со сдавливанием нервных стволов, кровеносных сосудов с нарушением их проходимости; встречается при коагулопатиях – гемофилии, передозировке антикоагулянтов, при тяжелом дефиците фактора VII и др. ); II. ПЕТЕХИАЛЬНО ПЯТНИСТЫЙ (микроциркуляторный) – вызывается количественным или качественным дефектом тромбоцитов (мелкие, размерами от точки до булавочной головки, кожные кровоизлияния – петехии; наряду с ними могут появляться синяки и кровоподтеки разных размеров – экхимозы. Наряду с кожными проявлениями для данного типа кровотточивости характерны геморрагии на слизистых оболочках; III. СМЕШАННЫЙ (микроциркуляторно гематомный) – имеет признаки гематомного и петехиально пятнистого типов. Преобладает микроциркуляторная кровоточивость; IV. ВАСКУЛИТНО ПУРПУРНЫЙ (поражения эндотелия сосудов воспалительного или иммунного характера – на коже нижних конечностей, внизу живота и туловища. Высыпания симметричные, ярко красного цвета); V. АНГИОМАТОЗНЫЙ (обусловлен кровотечением из мест, где имеются телеангиэктазии или ангиомы).
Типы кровоточивости: l l l I. ГЕМАТОМНЫЙ (массивные, глубокие, напряженные и очень болезненные кровоизлияния в крупные суставы, мышцы, подкожную и забрюшинную клетчатку, серозные оболочки со сдавливанием нервных стволов, кровеносных сосудов с нарушением их проходимости; встречается при коагулопатиях – гемофилии, передозировке антикоагулянтов, при тяжелом дефиците фактора VII и др. ); II. ПЕТЕХИАЛЬНО ПЯТНИСТЫЙ (микроциркуляторный) – вызывается количественным или качественным дефектом тромбоцитов (мелкие, размерами от точки до булавочной головки, кожные кровоизлияния – петехии; наряду с ними могут появляться синяки и кровоподтеки разных размеров – экхимозы. Наряду с кожными проявлениями для данного типа кровотточивости характерны геморрагии на слизистых оболочках; III. СМЕШАННЫЙ (микроциркуляторно гематомный) – имеет признаки гематомного и петехиально пятнистого типов. Преобладает микроциркуляторная кровоточивость; IV. ВАСКУЛИТНО ПУРПУРНЫЙ (поражения эндотелия сосудов воспалительного или иммунного характера – на коже нижних конечностей, внизу живота и туловища. Высыпания симметричные, ярко красного цвета); V. АНГИОМАТОЗНЫЙ (обусловлен кровотечением из мест, где имеются телеангиэктазии или ангиомы).
 Гемофилия l Гемофилия представляет собой генетическое заболевание, наследуемое по рецессивному типу, сцепленному с полом.
Гемофилия l Гемофилия представляет собой генетическое заболевание, наследуемое по рецессивному типу, сцепленному с полом.
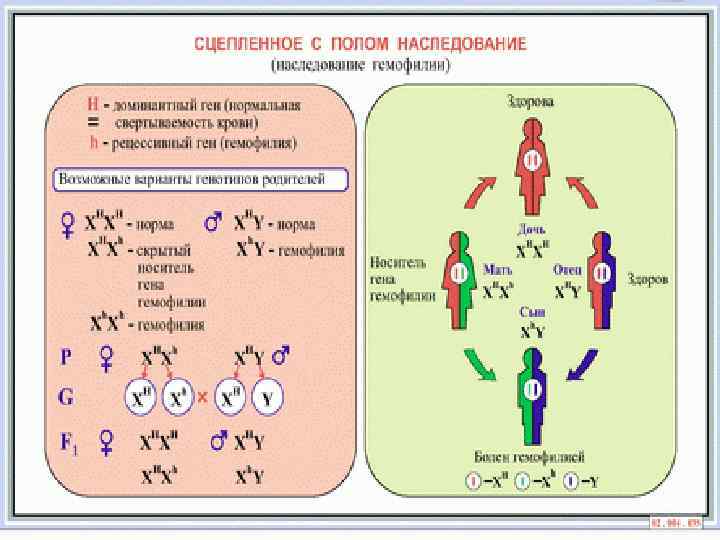
 локализуется в Х хромосоме,") Патогенез: Ген, ответственный за синтез одного из прокоагулянтов (антигемофилического глобулина) локализуется в Х хромосоме, вследствии чего гемофилией заболевают почти исключительно мужчины (половина сыновей этих женщин, имеющих ген гемофилии, имеют шансы родиться больными, при равной возможности получить от матери патологическую или нормальную Х хромосому, а половина дочерей стать передатчицами болезни). Заболевание женщины возможно в случае брака между больным гемофилией и женщиной носителем патологической хромосомы. Локализующийся в Х хромосоме ген гемофилии передается от больного гемофилией всем его дочерям, и они в дальнейшем передают этот ген своим потомкам. Все сыновья больного остаются здоровыми, так как получают единственную Х хромосому от здоровой матери. Основной механизм кровоточивости при гемофилии заключается в нарушении первой фазы свертывания крови — фазы образования тромбопластина (протромбиназы). Образование тромбопластина нарушено вследствии недостатка VIII фактора (антигемофилитического глобулина) или IX фактора (плаз менного компонента тромбопластина). При отсутствии или недостаточной активности этих факторов выпадает одно из звеньев последовательной активации плазменных факторов, приводящих к образованию тромбопластина. В зависимости от того, имеется ли недостаток VIII или IX факторов, различают 2 типа гемофилии. Заболевание, связанное с недостатком VIII фактора, обозначают гемофилией А, а при недостатке IX фактора говорят о гемофилии В. Частота гемофилии А составляет 85 90%, а гемофилии В 10 15% от всех гемофилии. Известна также гемофилия С, связанная с дефицитом XI фактора.
Патогенез: Ген, ответственный за синтез одного из прокоагулянтов (антигемофилического глобулина) локализуется в Х хромосоме, вследствии чего гемофилией заболевают почти исключительно мужчины (половина сыновей этих женщин, имеющих ген гемофилии, имеют шансы родиться больными, при равной возможности получить от матери патологическую или нормальную Х хромосому, а половина дочерей стать передатчицами болезни). Заболевание женщины возможно в случае брака между больным гемофилией и женщиной носителем патологической хромосомы. Локализующийся в Х хромосоме ген гемофилии передается от больного гемофилией всем его дочерям, и они в дальнейшем передают этот ген своим потомкам. Все сыновья больного остаются здоровыми, так как получают единственную Х хромосому от здоровой матери. Основной механизм кровоточивости при гемофилии заключается в нарушении первой фазы свертывания крови — фазы образования тромбопластина (протромбиназы). Образование тромбопластина нарушено вследствии недостатка VIII фактора (антигемофилитического глобулина) или IX фактора (плаз менного компонента тромбопластина). При отсутствии или недостаточной активности этих факторов выпадает одно из звеньев последовательной активации плазменных факторов, приводящих к образованию тромбопластина. В зависимости от того, имеется ли недостаток VIII или IX факторов, различают 2 типа гемофилии. Заболевание, связанное с недостатком VIII фактора, обозначают гемофилией А, а при недостатке IX фактора говорят о гемофилии В. Частота гемофилии А составляет 85 90%, а гемофилии В 10 15% от всех гемофилии. Известна также гемофилия С, связанная с дефицитом XI фактора.
 Клиническая картина: Гемофилия А и В клинически не отличаются друг от друга. Основным симптомом заболевания является повышенная кровоточивость, возникающая после уколов, порезов, ушибов, экстракции зубов и т. д. Иногда кровоточивость появляется после незначительных травм, что создает впечатление спонтанных кровотечений. Часто отмечаются обширные подкожные, межмышечные, субфасциальные и забрюшинные гематомы. При возникновении гематом происходит сдавление сосудов и нервов с развитием болевого синдрома и некроза окружающей ткани. Инфильтртруясь они могут быть причиной тяжелого сепсиса, при локализации в мягких тканях подчелюстной области, шеи, зева и глотки вызывают стеноз верхних дыхательных путей и асфиксию. Часто имеют место кровотечения из внутренних органов, в частности почечные кровотечения, при которых возможна закупорка почечных канальцев сгустками крови, что сопровождается приступами почечной колики. Реже возникают кишечные и легочные кровотечения. Желудочно кишечные кровотечения при гемофилии бывают спонтанными или обусловлены приемом некоторых лекарственных средств, вызывающих эрозию слизистой оболочки ЖКТ или обладающих антиагрегантными свойствами. Источником кровотечений могут быть язвы желудка и двенадцатиперстной кишки. Геморрагии в брыжейку, сальник, стенку кишки имитируют острые хирургические за болевания органов брюшной полости. Эффективность заместительной терапии служит диагностическим критерием в подобных случаях. Для гемофилии характерны длительные кровотечения при травмах и операциях, поэтому любое хирургическое вмешательство требуег введения антигемофильных препаратов. Кровоизлияния в головной и спинной мозг и их оболочки почти всегда связаны либо с травмами, либо употреблением дезагрегантов.
Клиническая картина: Гемофилия А и В клинически не отличаются друг от друга. Основным симптомом заболевания является повышенная кровоточивость, возникающая после уколов, порезов, ушибов, экстракции зубов и т. д. Иногда кровоточивость появляется после незначительных травм, что создает впечатление спонтанных кровотечений. Часто отмечаются обширные подкожные, межмышечные, субфасциальные и забрюшинные гематомы. При возникновении гематом происходит сдавление сосудов и нервов с развитием болевого синдрома и некроза окружающей ткани. Инфильтртруясь они могут быть причиной тяжелого сепсиса, при локализации в мягких тканях подчелюстной области, шеи, зева и глотки вызывают стеноз верхних дыхательных путей и асфиксию. Часто имеют место кровотечения из внутренних органов, в частности почечные кровотечения, при которых возможна закупорка почечных канальцев сгустками крови, что сопровождается приступами почечной колики. Реже возникают кишечные и легочные кровотечения. Желудочно кишечные кровотечения при гемофилии бывают спонтанными или обусловлены приемом некоторых лекарственных средств, вызывающих эрозию слизистой оболочки ЖКТ или обладающих антиагрегантными свойствами. Источником кровотечений могут быть язвы желудка и двенадцатиперстной кишки. Геморрагии в брыжейку, сальник, стенку кишки имитируют острые хирургические за болевания органов брюшной полости. Эффективность заместительной терапии служит диагностическим критерием в подобных случаях. Для гемофилии характерны длительные кровотечения при травмах и операциях, поэтому любое хирургическое вмешательство требуег введения антигемофильных препаратов. Кровоизлияния в головной и спинной мозг и их оболочки почти всегда связаны либо с травмами, либо употреблением дезагрегантов.
 Характерной особенностью гемофилии являются кровоизлияния в суставы гемартрозы, возникающие обычно после незначительных травм. Чаще других поражаются коленные, локтевые, голеностопные суставы. Клинически при свежих гемартрозах отмечается припухлость сустава, повышение местной температуры, болезненность и ограниченность движений. В связи с повторными кровоизлияниями возникают фиброзные изменения в суставах, контрактуры, деформации. Рентгенологически в полости сустава или суставной сумке выявляются тени, анатомическим субстратом которых являются обызвествленные кровяные сгустки. Кроме внутрисуставных гематом при гемофилии могут наблюдаться поднадкостничные гематомы, проявляющиеся в виде твердой припухлости. Кровоточивость при гемофилии обнаруживается уже на первом году жизни. С возрастом проявления кровоточивости становятся менее выраженными. В течении заболевания периоды кровоточивости сменяются периодами относительного благополучия. Существуют скрытые формы гемофилии, которые длительное время могут протекать бессимптомно и выявляются в связи с травмой или оперативными вмешательствами.
Характерной особенностью гемофилии являются кровоизлияния в суставы гемартрозы, возникающие обычно после незначительных травм. Чаще других поражаются коленные, локтевые, голеностопные суставы. Клинически при свежих гемартрозах отмечается припухлость сустава, повышение местной температуры, болезненность и ограниченность движений. В связи с повторными кровоизлияниями возникают фиброзные изменения в суставах, контрактуры, деформации. Рентгенологически в полости сустава или суставной сумке выявляются тени, анатомическим субстратом которых являются обызвествленные кровяные сгустки. Кроме внутрисуставных гематом при гемофилии могут наблюдаться поднадкостничные гематомы, проявляющиеся в виде твердой припухлости. Кровоточивость при гемофилии обнаруживается уже на первом году жизни. С возрастом проявления кровоточивости становятся менее выраженными. В течении заболевания периоды кровоточивости сменяются периодами относительного благополучия. Существуют скрытые формы гемофилии, которые длительное время могут протекать бессимптомно и выявляются в связи с травмой или оперативными вмешательствами.
 Диагностика: Диагноз гемофилии ставится на основании клинической картины (кровоточивость после мелких операций и травм, гемартрозы), наличия семейных случаев заболевания но мужской линии и данных лабораторных исследований. Важнейшим лабораторным тестом является значительное удлинение времени свертывания и времени рекальцификации плазмы, характеризующее на рушение в 1 фазегемокоагуляции. Время кровотечения, ретракция кровяного сгустка нормальны, пробы жгута и щипка при гемофилии отрицательные. Дефицит различных факторов свертывания и определение вида гемофилии проводят с помощью коррекционных проб. Выясняют, устраняется ли нарушение свертывания компонентами крови с заведомо известным недостатком того или иного фактора. Если дефект свертывания, выявленный у больного, устраняется только адсорбированной сульфатом бария (Ba. S 04) плазмой, в которой присутствует фактор VIII, но отсутствует фактор IX, то можно поставить диагноз гемофилии А. Если дефект исправляется только с помощью нормальной сыворотки, в которой присутствует фактор IX, но отсутствует фактор VIII, то ставят диагноз гемофилии В. Вид гемофилии может быть определен также с помощью тестов смешивания. К плазме обследуемого добавляют последовательно в разных пробирках образцы плазмы больных с заведомо известной формой гемофилии, т. е. , резким снижением уровней факторов VIII и IX, определяют время свертывания (или время рекальцификации). При смешивании образцов с дефицитом одного фактора коррекции времени свертывания не происходит, в то время, как смешива ние образцов с дефицитом разных факторов свертывания ведет к взаимной коррекции этих факторов и нормализации процесса свертывания. Диагностика завершается количественным определением уровня дефицитного фактора. Присутствие в плазме крови больного ингибитора фактора VIII : к доказывается с помощью гемагглютинационного теста (проба Кумбса) с использованием антител против фактора VIII : к или теста, основанного на способности плазмы больного при добавлении к нормальной плазме удлинять парциальное тромбопластиновое время свертывания последней. Дифференциальную диагностику следует проводить с болезнью Виллеб ранда и гемофилией В. В основе болезни Виллебранда наследственного геморрагического диатеза с аутосомно доминмнтным типом передачи лежит нарушение синтеза эндотелием сосудистой стенки крупномолекулярного компонента фактора VIII или ристоцетин кофактора. Болеют лица обоего пола. Выраженность геморрагического синдрома при болезни Виллебранда разная от сравнительно легких форм до тяжелых
Диагностика: Диагноз гемофилии ставится на основании клинической картины (кровоточивость после мелких операций и травм, гемартрозы), наличия семейных случаев заболевания но мужской линии и данных лабораторных исследований. Важнейшим лабораторным тестом является значительное удлинение времени свертывания и времени рекальцификации плазмы, характеризующее на рушение в 1 фазегемокоагуляции. Время кровотечения, ретракция кровяного сгустка нормальны, пробы жгута и щипка при гемофилии отрицательные. Дефицит различных факторов свертывания и определение вида гемофилии проводят с помощью коррекционных проб. Выясняют, устраняется ли нарушение свертывания компонентами крови с заведомо известным недостатком того или иного фактора. Если дефект свертывания, выявленный у больного, устраняется только адсорбированной сульфатом бария (Ba. S 04) плазмой, в которой присутствует фактор VIII, но отсутствует фактор IX, то можно поставить диагноз гемофилии А. Если дефект исправляется только с помощью нормальной сыворотки, в которой присутствует фактор IX, но отсутствует фактор VIII, то ставят диагноз гемофилии В. Вид гемофилии может быть определен также с помощью тестов смешивания. К плазме обследуемого добавляют последовательно в разных пробирках образцы плазмы больных с заведомо известной формой гемофилии, т. е. , резким снижением уровней факторов VIII и IX, определяют время свертывания (или время рекальцификации). При смешивании образцов с дефицитом одного фактора коррекции времени свертывания не происходит, в то время, как смешива ние образцов с дефицитом разных факторов свертывания ведет к взаимной коррекции этих факторов и нормализации процесса свертывания. Диагностика завершается количественным определением уровня дефицитного фактора. Присутствие в плазме крови больного ингибитора фактора VIII : к доказывается с помощью гемагглютинационного теста (проба Кумбса) с использованием антител против фактора VIII : к или теста, основанного на способности плазмы больного при добавлении к нормальной плазме удлинять парциальное тромбопластиновое время свертывания последней. Дифференциальную диагностику следует проводить с болезнью Виллеб ранда и гемофилией В. В основе болезни Виллебранда наследственного геморрагического диатеза с аутосомно доминмнтным типом передачи лежит нарушение синтеза эндотелием сосудистой стенки крупномолекулярного компонента фактора VIII или ристоцетин кофактора. Болеют лица обоего пола. Выраженность геморрагического синдрома при болезни Виллебранда разная от сравнительно легких форм до тяжелых
 Наиболее характерны носовые кровотечения, подкожные и внутрикожные геморрагии, но может наблюдаться и гематомный тип кровоточивости, как у больных гемофилией. Диагноз при классическом варианте болезни Виллебранда устанавливают на основании следующих признаков: значительного увеличения времени кровотечения, снижения адгезии тромбоцитов к стеклу и коллагену, а также ристоцидин агрегации, для которой также требуется УШ: ФВ, при нормальных других видах агрегации (под влиянием АДФ, адреналина, арахидоновой кислоты). Кроме того отмечается снижение коагуляционной активности фактора VIII (уровня фактора VIII: к), устраняемого переливанием не только нормальной плазмы, но и плазмы больных гемофилией с постепенным, через 4 8 ч, нарастанием активности фактора VIII : к (из за способности фактора Виллебранда, нормально присутствующего в переливаемой плазме, стимулировать синтез фактора VIII : к). Подтверждается диагноз определением уровня фактора Виллебранда в плазме больного по влиянию разных ее разведений на ристоцедин агрегацию отмытых нормальных тромбоцитов и радиоиммунньш методом. Гемофилия В (болезнь Кристмаса) наследственный геморрагический диатез, обусловленный дефицитом активности фактора IX (плазменного ком понентатромбопластина). Передается по рецессивному, цсепленному с X хромосомой типу. Структурный ген фактора IX не связан с фактором VIII, гак как располагается на другом конце Х хромосомы. Ген фактора мутирует в 7 10 раз реже, чем ген фактора VIII, поэтому на долю болезни Кристмаса приходиться 8 15% всех случаев гемофилии. У большинства больных гемофилией В в плазме отсутствует антиген фактора IX, иммунные формы встречаются редко. Клиническая симптоматика, характер течения, возможные осложнения при болезни Кристмаса идентичны таковым при гемофилии А. В дифференциальной диагностике важно учитывать данные лабораторных исследований и тестов корреляции. Необходимо количественное определение фактора для оценки тяжести болезни
Наиболее характерны носовые кровотечения, подкожные и внутрикожные геморрагии, но может наблюдаться и гематомный тип кровоточивости, как у больных гемофилией. Диагноз при классическом варианте болезни Виллебранда устанавливают на основании следующих признаков: значительного увеличения времени кровотечения, снижения адгезии тромбоцитов к стеклу и коллагену, а также ристоцидин агрегации, для которой также требуется УШ: ФВ, при нормальных других видах агрегации (под влиянием АДФ, адреналина, арахидоновой кислоты). Кроме того отмечается снижение коагуляционной активности фактора VIII (уровня фактора VIII: к), устраняемого переливанием не только нормальной плазмы, но и плазмы больных гемофилией с постепенным, через 4 8 ч, нарастанием активности фактора VIII : к (из за способности фактора Виллебранда, нормально присутствующего в переливаемой плазме, стимулировать синтез фактора VIII : к). Подтверждается диагноз определением уровня фактора Виллебранда в плазме больного по влиянию разных ее разведений на ристоцедин агрегацию отмытых нормальных тромбоцитов и радиоиммунньш методом. Гемофилия В (болезнь Кристмаса) наследственный геморрагический диатез, обусловленный дефицитом активности фактора IX (плазменного ком понентатромбопластина). Передается по рецессивному, цсепленному с X хромосомой типу. Структурный ген фактора IX не связан с фактором VIII, гак как располагается на другом конце Х хромосомы. Ген фактора мутирует в 7 10 раз реже, чем ген фактора VIII, поэтому на долю болезни Кристмаса приходиться 8 15% всех случаев гемофилии. У большинства больных гемофилией В в плазме отсутствует антиген фактора IX, иммунные формы встречаются редко. Клиническая симптоматика, характер течения, возможные осложнения при болезни Кристмаса идентичны таковым при гемофилии А. В дифференциальной диагностике важно учитывать данные лабораторных исследований и тестов корреляции. Необходимо количественное определение фактора для оценки тяжести болезни
 Лечение: Базисной терапией гемофилических геморрагии любой локализации и любого генеза является внутривенное введение достаточных доз гемопрепаратов, содержащих фактор VIII. Используют трансфузии свежеполученной крови или прямые гемотрансфузии (при отсутствии других антигемофиль ных препаратов), введения антигемофильной плазмы, концентратов фактора VIII. Мать больного гемофилией не должна использоваться в качестве донора. Наиболее эффективны концентраты фактора VIII криопреципитат и другие препараты, среди которых имеются стандартизованные сухие концентраты, которые могут храниться при комнатной температуре и удобны для транспортировки. Антигемофильные препараты вводят внутривенно струйно, инфузии повторяют через 8 12 ч (период полужизни фактора VIII). При контроле за трансфузионной терапией ориентируются на количественное определение уровня фактора VIII в плазме крови. С целью купирования умеренных геморрагии необходимо повысить уровень в плазме фактора VIII до 15 20%, для чего вводят антигемофильную плазму в дозе 10 15 мл/кг; более тяжелые кровотечения требуют поддержания уровня фактора VIII выше 30%, что достигается введением криопреципитата или других концентратов фактора VIII по 20 30 ед/кг и выше. При наружных геморрагиях из участков поврежденной кожи и слизистых оболочек наряду с трансфузионной терапией применяют местные воздействия обработку кровоточащих участков тромбопластином, тромбином, охлажденной е аминоканроновой кислотой (5 6% раствор). Осумкованные гематомы удаляют хирургически. Одновременно проводят интенсивную терапию концентратами антигемофилических факторов. Желудочно кишечные кровотечения купируют большими дозами антигемофилическогоконцентрата в сочетании с е аминокапроновой кислотой. В комплексном лечении поражений опорно двигательного аппарата используют рентгенотерапию, физиотерапевтические и бальнеологические методы лечения, синовэктомию, применяют ортопедические аппараты, ахилопластику. Для купирования кровотечений у больных с ингибиторной формой гемофилии введение больших доз концентрата фактора V 1 I 1 и свежей антигемофильной плазмы сочетают с плазмаферезом или вводят концентраты факторов IX, X, И ( «лечение в обход» , при котором, однако, возникает опасность тромбозов).
Лечение: Базисной терапией гемофилических геморрагии любой локализации и любого генеза является внутривенное введение достаточных доз гемопрепаратов, содержащих фактор VIII. Используют трансфузии свежеполученной крови или прямые гемотрансфузии (при отсутствии других антигемофиль ных препаратов), введения антигемофильной плазмы, концентратов фактора VIII. Мать больного гемофилией не должна использоваться в качестве донора. Наиболее эффективны концентраты фактора VIII криопреципитат и другие препараты, среди которых имеются стандартизованные сухие концентраты, которые могут храниться при комнатной температуре и удобны для транспортировки. Антигемофильные препараты вводят внутривенно струйно, инфузии повторяют через 8 12 ч (период полужизни фактора VIII). При контроле за трансфузионной терапией ориентируются на количественное определение уровня фактора VIII в плазме крови. С целью купирования умеренных геморрагии необходимо повысить уровень в плазме фактора VIII до 15 20%, для чего вводят антигемофильную плазму в дозе 10 15 мл/кг; более тяжелые кровотечения требуют поддержания уровня фактора VIII выше 30%, что достигается введением криопреципитата или других концентратов фактора VIII по 20 30 ед/кг и выше. При наружных геморрагиях из участков поврежденной кожи и слизистых оболочек наряду с трансфузионной терапией применяют местные воздействия обработку кровоточащих участков тромбопластином, тромбином, охлажденной е аминоканроновой кислотой (5 6% раствор). Осумкованные гематомы удаляют хирургически. Одновременно проводят интенсивную терапию концентратами антигемофилических факторов. Желудочно кишечные кровотечения купируют большими дозами антигемофилическогоконцентрата в сочетании с е аминокапроновой кислотой. В комплексном лечении поражений опорно двигательного аппарата используют рентгенотерапию, физиотерапевтические и бальнеологические методы лечения, синовэктомию, применяют ортопедические аппараты, ахилопластику. Для купирования кровотечений у больных с ингибиторной формой гемофилии введение больших доз концентрата фактора V 1 I 1 и свежей антигемофильной плазмы сочетают с плазмаферезом или вводят концентраты факторов IX, X, И ( «лечение в обход» , при котором, однако, возникает опасность тромбозов).
 Массивная трансфузионная терапия, проводимая больным гемофилией, ведет к сенсибилизации пациентов, создает риск заражения вирусным гепатита В, а также лимфотропными вирусами, провоцирует гемолитическую анемию. При значительных, угрожающих жизни кровотечениях переливают 1 2 л, а при менее выраженной кровоточивости 250 500 мл плазмы. Следует иметь в виду, что антигемофилический глобулин нестоек и при хранении исчезает, в связи с чем необходимо переливать только свежезаготовленную плазму со сроком хранения не более 24. В настоящее время применяется выделенный из плазмы донора очищенный препарат антигемофилического глобулина, обладающего в 20 раз большей активностью, чем свежая плазма. Еще большей активностью обладает выделенный из замороженной плазмы криопротеин. В случае необходимости оперативного вмешательства у больных гемофилией показано переливание свежей крови или плазмы не менее 500 мл перед операцией и 500 1000 мл во время операции. При свежих гемартрозах применяют аспирацию крови из полости сустава, давящую повязку, иммобилизацию сустава. Некоторый эффект дают глюкокортикоидные гормоны (преднизолон в дозе 25 30 мг), снимающий отек периартикулярной ткани. При хронических гемофилических гемартрозах проводят рентгенотерапию пораженных суставов. В качестве местных кровоостанавливающих средств применяют фибриновую губку с тромбином, препараты змеиного яда. Профилактика гемофилических кровотечений заключается в предупреждении с раннего детского возраста всевозможных травм и повреждений. Все больные должны находиться под постоянным диспансерным наблюдение
Массивная трансфузионная терапия, проводимая больным гемофилией, ведет к сенсибилизации пациентов, создает риск заражения вирусным гепатита В, а также лимфотропными вирусами, провоцирует гемолитическую анемию. При значительных, угрожающих жизни кровотечениях переливают 1 2 л, а при менее выраженной кровоточивости 250 500 мл плазмы. Следует иметь в виду, что антигемофилический глобулин нестоек и при хранении исчезает, в связи с чем необходимо переливать только свежезаготовленную плазму со сроком хранения не более 24. В настоящее время применяется выделенный из плазмы донора очищенный препарат антигемофилического глобулина, обладающего в 20 раз большей активностью, чем свежая плазма. Еще большей активностью обладает выделенный из замороженной плазмы криопротеин. В случае необходимости оперативного вмешательства у больных гемофилией показано переливание свежей крови или плазмы не менее 500 мл перед операцией и 500 1000 мл во время операции. При свежих гемартрозах применяют аспирацию крови из полости сустава, давящую повязку, иммобилизацию сустава. Некоторый эффект дают глюкокортикоидные гормоны (преднизолон в дозе 25 30 мг), снимающий отек периартикулярной ткани. При хронических гемофилических гемартрозах проводят рентгенотерапию пораженных суставов. В качестве местных кровоостанавливающих средств применяют фибриновую губку с тромбином, препараты змеиного яда. Профилактика гемофилических кровотечений заключается в предупреждении с раннего детского возраста всевозможных травм и повреждений. Все больные должны находиться под постоянным диспансерным наблюдение
 К болезни Верльгофа принято относить те случаи тромбоцитопенических пурпур,") Болезнь Верльгофа (идиопатическая тромбоцитопеническая пурпура) К болезни Верльгофа принято относить те случаи тромбоцитопенических пурпур, при которых не удается выявить определенного этиологического фактора (идиопатическая тромбоцитопеническая пурпура). Она не связана с каким либо предшествующим заболеванием (основным), лекарственными или токсическими воздействиями. Патогенез: Выделяют две формы заболевания иммунную и неиммунную. В патогенезе основную роль отводят иммунным механизмам. При иммунных формах основной патогенетический механизм тромбоцитопении заключается в агглютинации тромбоцитов антителами. Кроме того, антитела могут влиять как ингибиторы на процесс отшнуровки кровяных пластинок мегакариоцитами костного мозга. В эту группу не входят лекарственные иммуноаллергические тромбоцитопении, при которых антитела образуются при воздействии различных лекарственных препаратов. Значение иммунных механизмов подтверждается возможностью переноса болезни индуцирования тромбоцитопении у здоровых лиц после переливания им плазмы крови больных с тромбоцитопенической пурпурой. Плазменные факторы, вызывающие снижение уровня тромбоцитов у здоровых, представляют собой поликлональные иммуноглобулины класса G, которые чаще бывают неполными антителами, адсорбирующимися на поверх ности тромбоцитов, в связи с чем их определение сопряжено со значительными методическими трудностями. Покрытые антителами тромбоциты удаляются из крови селезенкой, реже печенью; при гистологическом исследовании в макрофагах селезенки наблюдается большое количество тромбоцитов на разных стадиях разрушения. Усиление деструкции тромбоцитов (продолжительность жизни пластинок укорочена до нескольких часов вместо 8 10 дней в норме) влечет за собой компенсаторное увеличение их продукции в костном мозге, при этом появляются молодые формы мегакариоцитов. В тех случаях, когда действие антител направлено против антигенов мегакариоцитов, возможно нарушение продукции тромбоцитов.
Болезнь Верльгофа (идиопатическая тромбоцитопеническая пурпура) К болезни Верльгофа принято относить те случаи тромбоцитопенических пурпур, при которых не удается выявить определенного этиологического фактора (идиопатическая тромбоцитопеническая пурпура). Она не связана с каким либо предшествующим заболеванием (основным), лекарственными или токсическими воздействиями. Патогенез: Выделяют две формы заболевания иммунную и неиммунную. В патогенезе основную роль отводят иммунным механизмам. При иммунных формах основной патогенетический механизм тромбоцитопении заключается в агглютинации тромбоцитов антителами. Кроме того, антитела могут влиять как ингибиторы на процесс отшнуровки кровяных пластинок мегакариоцитами костного мозга. В эту группу не входят лекарственные иммуноаллергические тромбоцитопении, при которых антитела образуются при воздействии различных лекарственных препаратов. Значение иммунных механизмов подтверждается возможностью переноса болезни индуцирования тромбоцитопении у здоровых лиц после переливания им плазмы крови больных с тромбоцитопенической пурпурой. Плазменные факторы, вызывающие снижение уровня тромбоцитов у здоровых, представляют собой поликлональные иммуноглобулины класса G, которые чаще бывают неполными антителами, адсорбирующимися на поверх ности тромбоцитов, в связи с чем их определение сопряжено со значительными методическими трудностями. Покрытые антителами тромбоциты удаляются из крови селезенкой, реже печенью; при гистологическом исследовании в макрофагах селезенки наблюдается большое количество тромбоцитов на разных стадиях разрушения. Усиление деструкции тромбоцитов (продолжительность жизни пластинок укорочена до нескольких часов вместо 8 10 дней в норме) влечет за собой компенсаторное увеличение их продукции в костном мозге, при этом появляются молодые формы мегакариоцитов. В тех случаях, когда действие антител направлено против антигенов мегакариоцитов, возможно нарушение продукции тромбоцитов.
 Этиология: Этиология неиммунных форм заболевания связана, как полагают, с дефицитом «тромбоцитолоэтического фактора» , необходимого для нормальной дифференцировки и отшнуровки тромбоцитов. Клиническая картина: Начало заболевания во всех случаях острой формы у детей и части больных хронической формой бывает острым, при постепенном (исподволь) начале болезнь иногда обнаруживают случайно при анализе крови. Для геморрагического синдрома при тромбоцитопенической пурпуре характерны кровоизлияния в кожу в виде кровоподтеков и синяков на конечностях, туловище, а также лице, в конъюктиве глаза (предположить возможность у больного церебрального кровотечения позволяет обнаружение кровоизлияний в конъюктиве, вокруг глаз в сочетании с сильной головной болью), наблюдаются мелкоточечная (петехиальная) сыпь на нижних конечностях, иногда распространенная пурпура с экхимозами. Как правило, кровотечения возникают под влиянием незначительных травм. Кожные геморрагии локализуются чаще всего на передней поверхности туловища и конечностей. В местах инъекции появляются крупные кровоизлияния. В зависимости от давности, кровоизлияние на коже приобретает различную окраску. Характерным признаком заболевания являются кровотечения из слизистых оболочек. Наиболее часто наблюдаются носовые кровотечения, принимающие иногда профузный характер и приводящий к выраженной анемизации больных. Кровотечения из десен, вызываемые травмой, в частности экстракцией зуба, могут достигать большой интенсивности. У женщин на первый план в клинической картине выступают маточные кровотечения в виде меноррагий. Могут наблюдаться кровоизлияния в яичник, имитирующие картину внематочной беременности.
Этиология: Этиология неиммунных форм заболевания связана, как полагают, с дефицитом «тромбоцитолоэтического фактора» , необходимого для нормальной дифференцировки и отшнуровки тромбоцитов. Клиническая картина: Начало заболевания во всех случаях острой формы у детей и части больных хронической формой бывает острым, при постепенном (исподволь) начале болезнь иногда обнаруживают случайно при анализе крови. Для геморрагического синдрома при тромбоцитопенической пурпуре характерны кровоизлияния в кожу в виде кровоподтеков и синяков на конечностях, туловище, а также лице, в конъюктиве глаза (предположить возможность у больного церебрального кровотечения позволяет обнаружение кровоизлияний в конъюктиве, вокруг глаз в сочетании с сильной головной болью), наблюдаются мелкоточечная (петехиальная) сыпь на нижних конечностях, иногда распространенная пурпура с экхимозами. Как правило, кровотечения возникают под влиянием незначительных травм. Кожные геморрагии локализуются чаще всего на передней поверхности туловища и конечностей. В местах инъекции появляются крупные кровоизлияния. В зависимости от давности, кровоизлияние на коже приобретает различную окраску. Характерным признаком заболевания являются кровотечения из слизистых оболочек. Наиболее часто наблюдаются носовые кровотечения, принимающие иногда профузный характер и приводящий к выраженной анемизации больных. Кровотечения из десен, вызываемые травмой, в частности экстракцией зуба, могут достигать большой интенсивности. У женщин на первый план в клинической картине выступают маточные кровотечения в виде меноррагий. Могут наблюдаться кровоизлияния в яичник, имитирующие картину внематочной беременности.
 В некоторых случаях болезнь Верльгофа выявляется у девочек с появлением первых менструаций. Реже возникают кровотечения из внутренних органов: желудочно кишечные, почечные, кровоизлияния в полости. Острая форма чаше всего является следствием аутоиммунной тромбоцитопении. Хроническая форма характеризуется циклическим течением (сменой рецидивов и ремиссий). Рецидивы при болезни Верльгофа возникают обычно под влиянием интеркуррентных инфекций, травм, операций, беременности. При значительных кровонотерях возникает анемия нормохромного типа. В связи с длительными и повторными кровотечениями развивается гипохромная анемия железодефицитного характера. Со стороны белой крови при острых кровопотерях отмечается нейтрофильный лейкоцитоз. Количество тромбоцитов снижено. Особенно выражена тромбоцитопения в период кровотечений. Иногда в препарате обнаруживаются единичные тромбоциты или их количество уменьшено до нуля. В период ремиссий число тромбоцитов повышается, но не достигаег нормы. Морфологически тромбоциты характеризуются большой величиной, атипичной формой и скудной специфической зернистостью. В костном мозге характерным признаком является гиперплазия мегакариоцитарного ростка с увеличением молодых форм мегакариоцитов. Отшнуровка тромбоцитов от мегакариоцитов нарушена или отсутствует. Реакция кровяного сгустка на высоте тромбоцитопении не выявляется т. е. , сыворотка совершенно не отделяется. В период ремиссии ретракция кровяного сгустка может нормализоваться. Продолжительность кровотечения резко удлинена, свертывание крови обычно нормально. Симптомы щипка и жгута в период рецидивов положительные, в фазу ремиссии отрицательные.
В некоторых случаях болезнь Верльгофа выявляется у девочек с появлением первых менструаций. Реже возникают кровотечения из внутренних органов: желудочно кишечные, почечные, кровоизлияния в полости. Острая форма чаше всего является следствием аутоиммунной тромбоцитопении. Хроническая форма характеризуется циклическим течением (сменой рецидивов и ремиссий). Рецидивы при болезни Верльгофа возникают обычно под влиянием интеркуррентных инфекций, травм, операций, беременности. При значительных кровонотерях возникает анемия нормохромного типа. В связи с длительными и повторными кровотечениями развивается гипохромная анемия железодефицитного характера. Со стороны белой крови при острых кровопотерях отмечается нейтрофильный лейкоцитоз. Количество тромбоцитов снижено. Особенно выражена тромбоцитопения в период кровотечений. Иногда в препарате обнаруживаются единичные тромбоциты или их количество уменьшено до нуля. В период ремиссий число тромбоцитов повышается, но не достигаег нормы. Морфологически тромбоциты характеризуются большой величиной, атипичной формой и скудной специфической зернистостью. В костном мозге характерным признаком является гиперплазия мегакариоцитарного ростка с увеличением молодых форм мегакариоцитов. Отшнуровка тромбоцитов от мегакариоцитов нарушена или отсутствует. Реакция кровяного сгустка на высоте тромбоцитопении не выявляется т. е. , сыворотка совершенно не отделяется. В период ремиссии ретракция кровяного сгустка может нормализоваться. Продолжительность кровотечения резко удлинена, свертывание крови обычно нормально. Симптомы щипка и жгута в период рецидивов положительные, в фазу ремиссии отрицательные.
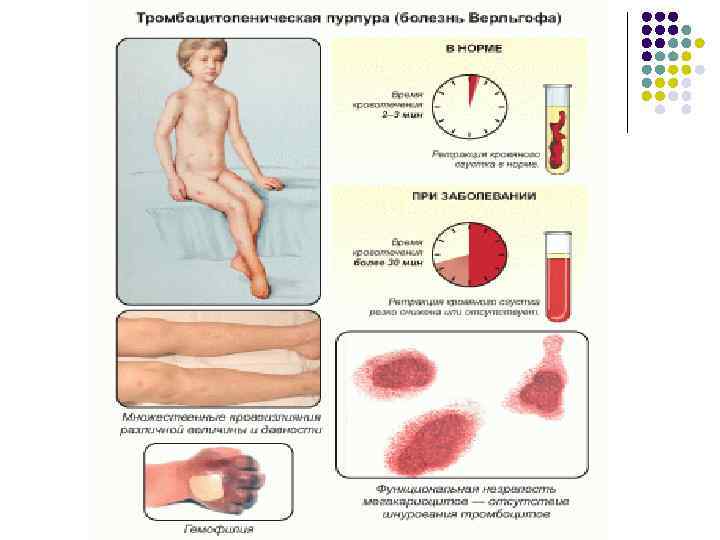
 Лабораторные данные Отмечается снижение количества тромбоцитов в разной степени, иногда вплоть до полного их исчезновения в крови. Геморрагический синдром редко наблюдается при уровне тромбоцитов более 50 10 /л. Могут обнаруживаться морфологические изменения тромбоцитов, увеличение числа крупных форм с уменьшением грануломера ( «голубые» пластинки), пойкилоцитоз, нарушения функций адгезии, АДФ и коллагенагрегации. У некоторых больных аутоиммунная тромбоцитопения сочетается с аутоиммунной анемией, но может развиться и железодефицитная анемия; уровень ретикулоцитов зависит от величины кровопотери и выраженности гемолиза. У части больных наблюдаются небольшой лейкоцитоз и эозинофилия, иногда аутоиммунная лейкопения. Время кровотечения обычно увеличено, коагуляционные пробы (время свертывания крови, парциальное тромбопластиновое время и др. ) не изменены. В костном мозге обнаруживают нормальное или чаще повышенное число мегакариоцитов со сдвигом в сторону молодых форм, что отражает активную продукцию тромбоцитов. При гемолизе или кровопотерях наблюдается увеличение числа клеток эритроцитарного ряда. У 60% больных детей в случае острой формы болезни выздоровление на ступает в течении 4 6 нед, у остальных заболевание длится дольше до 3 6 мес, но также заканчивается выздоровлением. Хроническая форма отличается затяжным или рецидивирующим течением. Дети, рожденные от женщин, страдающих аутоиммунноц тромбоцитопенией, нередко имеют тромбоцитопению и геморрагический синдром в результате разрушения тромбоцитов антителами матери, проникшими через плаценту в организм ребенка.
Лабораторные данные Отмечается снижение количества тромбоцитов в разной степени, иногда вплоть до полного их исчезновения в крови. Геморрагический синдром редко наблюдается при уровне тромбоцитов более 50 10 /л. Могут обнаруживаться морфологические изменения тромбоцитов, увеличение числа крупных форм с уменьшением грануломера ( «голубые» пластинки), пойкилоцитоз, нарушения функций адгезии, АДФ и коллагенагрегации. У некоторых больных аутоиммунная тромбоцитопения сочетается с аутоиммунной анемией, но может развиться и железодефицитная анемия; уровень ретикулоцитов зависит от величины кровопотери и выраженности гемолиза. У части больных наблюдаются небольшой лейкоцитоз и эозинофилия, иногда аутоиммунная лейкопения. Время кровотечения обычно увеличено, коагуляционные пробы (время свертывания крови, парциальное тромбопластиновое время и др. ) не изменены. В костном мозге обнаруживают нормальное или чаще повышенное число мегакариоцитов со сдвигом в сторону молодых форм, что отражает активную продукцию тромбоцитов. При гемолизе или кровопотерях наблюдается увеличение числа клеток эритроцитарного ряда. У 60% больных детей в случае острой формы болезни выздоровление на ступает в течении 4 6 нед, у остальных заболевание длится дольше до 3 6 мес, но также заканчивается выздоровлением. Хроническая форма отличается затяжным или рецидивирующим течением. Дети, рожденные от женщин, страдающих аутоиммунноц тромбоцитопенией, нередко имеют тромбоцитопению и геморрагический синдром в результате разрушения тромбоцитов антителами матери, проникшими через плаценту в организм ребенка.
 Диагностика: Диагноз основывается на клинических и лабораторных данных при исключении наследственных форм тромбоцитопении, симптоматических аутоиммунных тромбоцитопении, связанных с СКВ, хроническим активным гепатитом, хроническим лимфолейкозом, а также ДВС синдрома, тромботической тромбоцитопенической пурпуры и некоторых других заболеваний, протекающих с тромбоцитопенией. Для подтверждения аутоиммунной тромбоцитопении важное значение имеет обнаружение антитромбоцитарных антител с помощью различных методов, из которых наиболее информативен метод Диксона количественное определение антител на поверхности тромбоцитов (в норме 14 10 г/тромбоцит, при идеопатической тромбоцитопенической пурпуре 20 25010 ' /тромбоцит). Лечение: Лечение зависит от возраста больного, формы и тяжести болезни. Если у больного отсутствуют кровотечения и число тромбоцитов выше 20 10'/л, то терапию не проводят; в этих случаях, особенно у детей, оправдана выжидательная тактика. Коррекцию геморрагического синдрома в случаях острой и хронической форм болезни проводят с помощью глюкокортнкоидов, иногда для снижения уровня антител и иммунных комплексов назначают несколько сеансов плазмофереза и с целью временной блокады фагоцитирующих макрофагов введение гамма глобулина (400 мг/сут внутривенно в течение 5 дней). Преднизолон назначают в дозе 1 мг/кг в сутки в течение 3— 4 нед с последующим снижением дозы. Почти у половины больных такое лечение вызывает нормализацию числа тромбоцитов, однако у многих из нихоно вновь уменьшается после отмены препарата. У больных, чувствительных к преднизолону, существует большая вероятность эффективности сплеиэктомии. Показанием к спленэктамии является отсутствие стабильного эффекта от преднизолона. У 70% операция приводит к выздоровлению или стойкой ремис сии, причем увеличение количества тромбоцитов до нормы происходит уже в течении 2 нед после операции.
Диагностика: Диагноз основывается на клинических и лабораторных данных при исключении наследственных форм тромбоцитопении, симптоматических аутоиммунных тромбоцитопении, связанных с СКВ, хроническим активным гепатитом, хроническим лимфолейкозом, а также ДВС синдрома, тромботической тромбоцитопенической пурпуры и некоторых других заболеваний, протекающих с тромбоцитопенией. Для подтверждения аутоиммунной тромбоцитопении важное значение имеет обнаружение антитромбоцитарных антител с помощью различных методов, из которых наиболее информативен метод Диксона количественное определение антител на поверхности тромбоцитов (в норме 14 10 г/тромбоцит, при идеопатической тромбоцитопенической пурпуре 20 25010 ' /тромбоцит). Лечение: Лечение зависит от возраста больного, формы и тяжести болезни. Если у больного отсутствуют кровотечения и число тромбоцитов выше 20 10'/л, то терапию не проводят; в этих случаях, особенно у детей, оправдана выжидательная тактика. Коррекцию геморрагического синдрома в случаях острой и хронической форм болезни проводят с помощью глюкокортнкоидов, иногда для снижения уровня антител и иммунных комплексов назначают несколько сеансов плазмофереза и с целью временной блокады фагоцитирующих макрофагов введение гамма глобулина (400 мг/сут внутривенно в течение 5 дней). Преднизолон назначают в дозе 1 мг/кг в сутки в течение 3— 4 нед с последующим снижением дозы. Почти у половины больных такое лечение вызывает нормализацию числа тромбоцитов, однако у многих из нихоно вновь уменьшается после отмены препарата. У больных, чувствительных к преднизолону, существует большая вероятность эффективности сплеиэктомии. Показанием к спленэктамии является отсутствие стабильного эффекта от преднизолона. У 70% операция приводит к выздоровлению или стойкой ремис сии, причем увеличение количества тромбоцитов до нормы происходит уже в течении 2 нед после операции.
 Хороший эффект спленэктомии связывают с удалением основного органа образования антител и уменьшением массы клеток (макрофагов), фагоцитирующих и разрушающих тромбоциты. Больным, у которых спленэкгомия оказалась безуспешной, назначают цитостатическую терапию. При симптоматических аутоиммунных тромбоцитопениях (СКВ и другие системные заболевания соединительной ткани, хронический лимфолейкоз) активную терапию цитостатиками начинают раньше, спленэктомию производят при неэффективности этих средств и выраженных геморрагиях. Проводят, также, симптоматическое лечение; в последние годы с успехом применяют андроген диназол. Современное лечение уменьшило частоту осложнений; у большей части больных с хронической формой возможно выздоровление. Прогноз: Прогноз при хронических рецидивирующих формах благоприятный. В некоторых случаях кровотечения с возрастом ослабевают или вообще прекращаются. Менее благоприятны острые формы заболевания, при которых кровотечения могут достигать большой интенсивности. Трудоспособность в большинстве случаев сохранена.
Хороший эффект спленэктомии связывают с удалением основного органа образования антител и уменьшением массы клеток (макрофагов), фагоцитирующих и разрушающих тромбоциты. Больным, у которых спленэкгомия оказалась безуспешной, назначают цитостатическую терапию. При симптоматических аутоиммунных тромбоцитопениях (СКВ и другие системные заболевания соединительной ткани, хронический лимфолейкоз) активную терапию цитостатиками начинают раньше, спленэктомию производят при неэффективности этих средств и выраженных геморрагиях. Проводят, также, симптоматическое лечение; в последние годы с успехом применяют андроген диназол. Современное лечение уменьшило частоту осложнений; у большей части больных с хронической формой возможно выздоровление. Прогноз: Прогноз при хронических рецидивирующих формах благоприятный. В некоторых случаях кровотечения с возрастом ослабевают или вообще прекращаются. Менее благоприятны острые формы заболевания, при которых кровотечения могут достигать большой интенсивности. Трудоспособность в большинстве случаев сохранена.
 l Геморрагический васкулит (болезнь Шенлейна Геноха, анафилактоидная пурпура) системное заболевание,") Геморрагический васкулит (болезнь Шенлейна-Геноха) l Геморрагический васкулит (болезнь Шенлейна Геноха, анафилактоидная пурпура) системное заболевание, в основе которого, лежит поражение мелких сосудов капилляров и артериол.
Геморрагический васкулит (болезнь Шенлейна-Геноха) l Геморрагический васкулит (болезнь Шенлейна Геноха, анафилактоидная пурпура) системное заболевание, в основе которого, лежит поражение мелких сосудов капилляров и артериол.
") Распространенность: l Заболевают чаще мальчики и юноши, реже взрослые (одинаково часто мужчины и женщины) Клиническая картина: Заболевание обычно протекает с высокой температурой (до 39 40°). Одним из характерных признаков является сыпь на коже и слизистых оболочках (иногда отмечаются кожный зуд и небольшая отечность кожи). Часто кожным изменениям сопутствует суставной синдром, проявляющийся сильными болями в области крупных суставов (чаще коленных). Объективно отмечается припухлость суставов, болезненность, ограничение подвижности. Суставные явления носят нестойкий характер. Абдоминальный синдром при геморрагическом васкулите характеризуется приступообразными болями в животе, рвотой, с примесью крови, кровянистым стулом. Боли чаще всего локализуются вокруг пупка, реже в правой подвздошной области, имитируя картину аппендицита. Объективно отмечается вздутие, болезненность при пальпации, некоторое напряжение брюшной стенки. Тяжелым осложнением геморрагического васкулита является поражение почек, протекающего в виде диффузного гломерулонефрита с постепенным развитием признаков почечной недостаточности. В крови отмечается лейкоцитоз со сдвигом влево. Количество тромбоцитов в пределах нормы. Свертываемость крови и время кровотечения не изменены. Симптомы щипка и жгута непостоянны. Течение заболевания может быть острым и хронически рецидивирующим. В детском возрасте геморрагический васкулит протекает крайне тяжело в виде молниеносной пурпуры. Наряду с рецидивирующими формами существуют легкие, заканчивающиеся через некоторое время выздоровлением
Распространенность: l Заболевают чаще мальчики и юноши, реже взрослые (одинаково часто мужчины и женщины) Клиническая картина: Заболевание обычно протекает с высокой температурой (до 39 40°). Одним из характерных признаков является сыпь на коже и слизистых оболочках (иногда отмечаются кожный зуд и небольшая отечность кожи). Часто кожным изменениям сопутствует суставной синдром, проявляющийся сильными болями в области крупных суставов (чаще коленных). Объективно отмечается припухлость суставов, болезненность, ограничение подвижности. Суставные явления носят нестойкий характер. Абдоминальный синдром при геморрагическом васкулите характеризуется приступообразными болями в животе, рвотой, с примесью крови, кровянистым стулом. Боли чаще всего локализуются вокруг пупка, реже в правой подвздошной области, имитируя картину аппендицита. Объективно отмечается вздутие, болезненность при пальпации, некоторое напряжение брюшной стенки. Тяжелым осложнением геморрагического васкулита является поражение почек, протекающего в виде диффузного гломерулонефрита с постепенным развитием признаков почечной недостаточности. В крови отмечается лейкоцитоз со сдвигом влево. Количество тромбоцитов в пределах нормы. Свертываемость крови и время кровотечения не изменены. Симптомы щипка и жгута непостоянны. Течение заболевания может быть острым и хронически рецидивирующим. В детском возрасте геморрагический васкулит протекает крайне тяжело в виде молниеносной пурпуры. Наряду с рецидивирующими формами существуют легкие, заканчивающиеся через некоторое время выздоровлением
 Диагноз: Диагноз геморрагического васкулита ставится на основании высыпаний, сопровождающихся суставным и абдоминальным синдромами, отсутствия нарушений со стороны свертывающей системы При дифференциальной диагностике заболевания основные трудности встречаются в случаях абдоминального синдрома, имитирующего картину острого живота. В пользу геморрагического васкулита свидетельствуют схваткообразность болей, сопутствующие изменения кожи и суставов, отсутствие токсической зернистости в нейтрофилах. Прогноз: Прогноз при легких формах благоприятен. Ухудшает прогноз поражение почек, приводящее постепенно к развитию почечной недостаточности. Прогностически неблагоприятны молниеносные формы. Лечение: Строгий постельный режим. Гепаринотерапия в индивидуально подобранных дозах (100— 300 ед/кг/сут), подкожно, под строгим лабораторным контролем. Назначают криоплазму, курантил, десенсибилизирующие средства (тавегил, супрастин, димедрол по 0, 015— 0, 025 г 2 раза в день), аскорбиновую кислоту, рутин, витамин Р, в тяжелых случаях — преднизолон по 5— 15 мг в сутки. Диета с исключением индивидуальных сенсибилизирующих факторов.
Диагноз: Диагноз геморрагического васкулита ставится на основании высыпаний, сопровождающихся суставным и абдоминальным синдромами, отсутствия нарушений со стороны свертывающей системы При дифференциальной диагностике заболевания основные трудности встречаются в случаях абдоминального синдрома, имитирующего картину острого живота. В пользу геморрагического васкулита свидетельствуют схваткообразность болей, сопутствующие изменения кожи и суставов, отсутствие токсической зернистости в нейтрофилах. Прогноз: Прогноз при легких формах благоприятен. Ухудшает прогноз поражение почек, приводящее постепенно к развитию почечной недостаточности. Прогностически неблагоприятны молниеносные формы. Лечение: Строгий постельный режим. Гепаринотерапия в индивидуально подобранных дозах (100— 300 ед/кг/сут), подкожно, под строгим лабораторным контролем. Назначают криоплазму, курантил, десенсибилизирующие средства (тавегил, супрастин, димедрол по 0, 015— 0, 025 г 2 раза в день), аскорбиновую кислоту, рутин, витамин Р, в тяжелых случаях — преднизолон по 5— 15 мг в сутки. Диета с исключением индивидуальных сенсибилизирующих факторов.