

лекция 2011-аномалии развития.ppt
- Количество слайдов: 83

") Fortunio Liceti’s De Monstrorum causis natura (1665)
Fortunio Liceti’s De Monstrorum causis natura (1665)
 Уильям Гарвей месяц за месяцем следил за развитием эмбрионов косули и оставил одно из самых прекрасных описаний зародыша млекопитающих из всех когда-либо написанных "Уже давно я видел плод, размером с гороховый стручок, вырезанный из матки оленихи, который был полностью сформирован во всех своих частях, и я показывал его королю и королеве. Он плавал, балансировал и достигал совершенства в такой белой, абсолютно прозрачной и кристально чистой жидкости (как будто ее хранили в наичистейшем стеклянном сосуде), объемом с голубиное яйцо, где он помещался в своей собственной оболочке".
Уильям Гарвей месяц за месяцем следил за развитием эмбрионов косули и оставил одно из самых прекрасных описаний зародыша млекопитающих из всех когда-либо написанных "Уже давно я видел плод, размером с гороховый стручок, вырезанный из матки оленихи, который был полностью сформирован во всех своих частях, и я показывал его королю и королеве. Он плавал, балансировал и достигал совершенства в такой белой, абсолютно прозрачной и кристально чистой жидкости (как будто ее хранили в наичистейшем стеклянном сосуде), объемом с голубиное яйцо, где он помещался в своей собственной оболочке".
 Фредерик Рюйш (правильнее: Фредерик Рёйс Frederik") Урок анатомии Фредерика Рюйша (Ян ван Нек, 1683) Фредерик Рюйш (правильнее: Фредерик Рёйс Frederik Ruysch, 1638— 1731) — знаменитый голландский анатом, изучал медицину в Лейдене с 1665 г. — профессор анатомии, а с 1685 году и ботаники в Амстердаме
Урок анатомии Фредерика Рюйша (Ян ван Нек, 1683) Фредерик Рюйш (правильнее: Фредерик Рёйс Frederik Ruysch, 1638— 1731) — знаменитый голландский анатом, изучал медицину в Лейдене с 1665 г. — профессор анатомии, а с 1685 году и ботаники в Амстердаме
 В России всегда считалось, что уроды родятся от "действа дьявольского". Указ от 28 января 1704 года "о погребении умерших на третий день и о подтверждении повивальным бабкам, под смертной казнью, чтобы они младенцев, рождённых уродами не убивали и не "таили", а сообщали о них приходским священникам". Второй указ от 30 мая 1705 года адресован приходским священникам тотчас сообщать, если "которая череватая жена умрёт «, мёртвые тела детей и беременных женщин передавать в Анатомический театр. В указе Пётр 1 пытается научно объяснить происхождение уродства. Ещё Пётр 1 издал указы, которые предписывают жителям России за вознаграждение собирать и сдавать "всё, что зело старо и необыкновенно". Пётр 1 издал указ "О принесении родившихся уродов".
В России всегда считалось, что уроды родятся от "действа дьявольского". Указ от 28 января 1704 года "о погребении умерших на третий день и о подтверждении повивальным бабкам, под смертной казнью, чтобы они младенцев, рождённых уродами не убивали и не "таили", а сообщали о них приходским священникам". Второй указ от 30 мая 1705 года адресован приходским священникам тотчас сообщать, если "которая череватая жена умрёт «, мёртвые тела детей и беременных женщин передавать в Анатомический театр. В указе Пётр 1 пытается научно объяснить происхождение уродства. Ещё Пётр 1 издал указы, которые предписывают жителям России за вознаграждение собирать и сдавать "всё, что зело старо и необыкновенно". Пётр 1 издал указ "О принесении родившихся уродов".
 Великан Николай Буржуа Петр I 13 февраля 1718 года издал Именной указ «О порядке выдачи награждения за редкостные предметы» : «Понеже известно есть, что како в человеческой породе, так и в зверской и в птичьей случается, что родятся монстра, т. е. уроды, которые всегда во всех государствах сбираются для диковинки, чего для пред несколькими летами уже указ сказан, чтоб такие приносили, обещая платеж за оные, которых несколько уже и принесено. Однако ж в таком великом государстве может более быть; но таят невежды, чая, что такие уроды родятся от действа дьявольского, чрез ведовство и порчу; чему быть невозможно, ибо един творец всея твари бог, а не дьявол, которому ни над каким созданием власти нет, но от повреждения внутреннего и проч. . . За что давана будет довольная дача» .
Великан Николай Буржуа Петр I 13 февраля 1718 года издал Именной указ «О порядке выдачи награждения за редкостные предметы» : «Понеже известно есть, что како в человеческой породе, так и в зверской и в птичьей случается, что родятся монстра, т. е. уроды, которые всегда во всех государствах сбираются для диковинки, чего для пред несколькими летами уже указ сказан, чтоб такие приносили, обещая платеж за оные, которых несколько уже и принесено. Однако ж в таком великом государстве может более быть; но таят невежды, чая, что такие уроды родятся от действа дьявольского, чрез ведовство и порчу; чему быть невозможно, ибо един творец всея твари бог, а не дьявол, которому ни над каким созданием власти нет, но от повреждения внутреннего и проч. . . За что давана будет довольная дача» .
 Описал синдром Поланда (синдактилия в сочетании с агенезией малой грудной") Альфред ПОЛАНД (1822 -1872) Описал синдром Поланда (синдактилия в сочетании с агенезией малой грудной мышцы Пётр Андреевич ЗАГОРСКИЙ (1764 -1846) – русский анатом и физиолог, основатель русской анатомической школы. 1812 «Обозрение разнообразных человеческих уродств
Альфред ПОЛАНД (1822 -1872) Описал синдром Поланда (синдактилия в сочетании с агенезией малой грудной мышцы Пётр Андреевич ЗАГОРСКИЙ (1764 -1846) – русский анатом и физиолог, основатель русской анатомической школы. 1812 «Обозрение разнообразных человеческих уродств
 Описал синдром Дауна в 1866 под названием - синдром") Джон Лангдон ДАУН (1828 -1896) Описал синдром Дауна в 1866 под названием - синдром «монголизма» Ребёнок с характерными чертами, присущими синдрому Дауна (эпикантус, плоское лицо, открытый рот, увеличенный язык, маленький нос и т. д. )
Джон Лангдон ДАУН (1828 -1896) Описал синдром Дауна в 1866 под названием - синдром «монголизма» Ребёнок с характерными чертами, присущими синдрому Дауна (эпикантус, плоское лицо, открытый рот, увеличенный язык, маленький нос и т. д. )
, В 1896 сообщил о девочке с") Антонин Бернар Жан МАРФАН (Antonin-Bernard-Jean Marfan, 1858 -1942), В 1896 сообщил о девочке с астеничным телосложением, патологией скелета, сердца, глазных яблок Помимо триады Марфана, в других органах также наблюдаются изменения (бронхо-лёгочная система, кожа, твёрдая мозговая оболочка). (Болезнь Марфана, Marfan syndrome) — заболевание из группы наследственных коллагенопатий, заболеваний соединительной ткани человека. Частный случай дифференцированной дисплазии соединительной ткани человека. Наследственное заболевание, входит под номером 154700 в систему табуляции Мак. Кьюсика OMIM. Преимущественно эта болезнь наследуется по доминантному признаку и вызывается аномалией гена FBN 1 (15 хромосома), кодирующего белок фибрилин-1, являющийся компонентом внеклеточного матрикса
Антонин Бернар Жан МАРФАН (Antonin-Bernard-Jean Marfan, 1858 -1942), В 1896 сообщил о девочке с астеничным телосложением, патологией скелета, сердца, глазных яблок Помимо триады Марфана, в других органах также наблюдаются изменения (бронхо-лёгочная система, кожа, твёрдая мозговая оболочка). (Болезнь Марфана, Marfan syndrome) — заболевание из группы наследственных коллагенопатий, заболеваний соединительной ткани человека. Частный случай дифференцированной дисплазии соединительной ткани человека. Наследственное заболевание, входит под номером 154700 в систему табуляции Мак. Кьюсика OMIM. Преимущественно эта болезнь наследуется по доминантному признаку и вызывается аномалией гена FBN 1 (15 хромосома), кодирующего белок фибрилин-1, являющийся компонентом внеклеточного матрикса
 http: //www. ncbi. nlm. nih. gov/omim Проект «Менделевское наследование у человека» (англ. Mendelian Inheritance in Man, MIM) — медицинская база данных, в которой собирается информация об известных заболеваниях с генетическим компонентом и генах, ответственных за их развитие. Существуют две версии проекта. • Проект периодически издаётся в книжном варианте (текущее 12 -е издание). • Интернет-вариант базы данных OMIM (Online Mendelian Inheritance in Man) доступен через базу данных Entrez и является частью проекта «Образование» Национального центра биотехнологической информации NCBI
http: //www. ncbi. nlm. nih. gov/omim Проект «Менделевское наследование у человека» (англ. Mendelian Inheritance in Man, MIM) — медицинская база данных, в которой собирается информация об известных заболеваниях с генетическим компонентом и генах, ответственных за их развитие. Существуют две версии проекта. • Проект периодически издаётся в книжном варианте (текущее 12 -е издание). • Интернет-вариант базы данных OMIM (Online Mendelian Inheritance in Man) доступен через базу данных Entrez и является частью проекта «Образование» Национального центра биотехнологической информации NCBI
, В 1956 первая публикация о наследственных заболеваниях") Victor Almon Mc. Kusick (1921 – 2008), В 1956 первая публикация о наследственных заболеваниях Владимир Павлович Эфроимсон (1908 — 1989) , В 1964 монография о синдромальных формах наследственной патологии Birth defects original article series Publisher: National Foundation Other titles: Birth defects original article series, Birth defects From 1965 ISSN: 0547 -6844, OCLC: 1536497 Material type: Series Document type: Journal / Magazine / Newspaper
Victor Almon Mc. Kusick (1921 – 2008), В 1956 первая публикация о наследственных заболеваниях Владимир Павлович Эфроимсон (1908 — 1989) , В 1964 монография о синдромальных формах наследственной патологии Birth defects original article series Publisher: National Foundation Other titles: Birth defects original article series, Birth defects From 1965 ISSN: 0547 -6844, OCLC: 1536497 Material type: Series Document type: Journal / Magazine / Newspaper
 1925 год - первая монография на русском языке по клинической генетике Сергея Николаевича Давиденкова "Наследственные болезни нервной системы". 1934 год - публикация монографии С. Н. Давиденкова «Проблема полиморфизма наследственных болезней нервной системы» 1947 год - "появление книги С. Н. Давиденкова "Эволюционные и генетические проблемы в невропатологии" 1970 года - первое прекрасно иллюстрированное руководство-атлас по диагностике синдромов врожденных пороков развития Дэвида Смита (1926 -1981) - одного из пионеров в области синдромологии 1970 год - издание атласа Richard Goodman (1931 -1989) и Robert Gorlin - известных американских генетиков. Прекрасно иллюстрированное руководство ("Лицо при наследственных заболеваниях") по фенотипической диагностике синдромов, являющееся одной из настольных книг врача-генетика 1971 год - первая профессиональная монография на русском языке трех известных отечественных педиатров-профессоров "Наследственные болезни у детей" (Бадалян Л, О, , Таболин В. А. , Вельтищев Ю. А. , 1971). Долгие годы являлась настольной книгой педиатра и врача-генетика и до сих пор не утратила своего значения в качестве введения в специальность. 1971 год - первый на русском языке справочник по наследственным синдромам под редакцией известного невропатолога проф. Бадаляна Л. О. «Справочник по клинической генетике» .
1925 год - первая монография на русском языке по клинической генетике Сергея Николаевича Давиденкова "Наследственные болезни нервной системы". 1934 год - публикация монографии С. Н. Давиденкова «Проблема полиморфизма наследственных болезней нервной системы» 1947 год - "появление книги С. Н. Давиденкова "Эволюционные и генетические проблемы в невропатологии" 1970 года - первое прекрасно иллюстрированное руководство-атлас по диагностике синдромов врожденных пороков развития Дэвида Смита (1926 -1981) - одного из пионеров в области синдромологии 1970 год - издание атласа Richard Goodman (1931 -1989) и Robert Gorlin - известных американских генетиков. Прекрасно иллюстрированное руководство ("Лицо при наследственных заболеваниях") по фенотипической диагностике синдромов, являющееся одной из настольных книг врача-генетика 1971 год - первая профессиональная монография на русском языке трех известных отечественных педиатров-профессоров "Наследственные болезни у детей" (Бадалян Л, О, , Таболин В. А. , Вельтищев Ю. А. , 1971). Долгие годы являлась настольной книгой педиатра и врача-генетика и до сих пор не утратила своего значения в качестве введения в специальность. 1971 год - первый на русском языке справочник по наследственным синдромам под редакцией известного невропатолога проф. Бадаляна Л. О. «Справочник по клинической генетике» .
 1973 год - первое издание фундаментальной сводки по врожденным дефектам развития и синдромальным формам патологии "Вirth Defects Compendium". Современная база данных этой сводки полностью компьютизирована (Buyse, 1980) 1974 год - первое обстоятельное исследование этиологии выраженной умственной отсталости показавшее, что синдромальные формы врожденных пороков развития в структуре олигофрении составляют 29%, а метаболические "синдромы" - 6, 5% (Kaveggia et al. , 1974) 1977 год - первый клинический атлас хромосомных заболеваний человека французских исследователей Jean de Grouchy and Catherine Turleau (de Grouchy, Turleau, 1977). Прекрасно иллюстрованное руководство по синдромологии хромосомных заболеваний 1979 год - обширная публикация John M. Opitz - одного из ведущих американских генетиков, об основных понятиях синдромологии врожденных пороков развития (Opitz JM. , 1979) 1980 год - начало работ по созданию компьтерных баз данных по синдромологии. Одной из самых первых программ по синдромологии появилась австралийская система POSSUM 1987 год - публикация первого на русском языке иллюстрированного словаря по наследственным синдромам (Козлова С. И. , Семанова Е. , Демикова Н. С. , Блинникова О. Е. , 1987). 1992 год - начало выпуска специализированного журнала "Clinical Dysmorphology" (редакторы: Robert Winter, Dian Donnai, Michael Baraitser), в котором публикуются работы по всем аспектам синдромологии врожденных дефектов развития. 1993 год - публикация диагностического обзора множественных врожденных аномалий Robin M. Winter and Michel Baraitser, где кратко систематизированы более 400 "новых" синдромов описанных за последнии годы (Winter, Baraitser, 1993). В дальнейшем эта база данных стала известна как "Лондонская база данных по дисморфологии" (Winter, Baraitser, 1993, 1996).
1973 год - первое издание фундаментальной сводки по врожденным дефектам развития и синдромальным формам патологии "Вirth Defects Compendium". Современная база данных этой сводки полностью компьютизирована (Buyse, 1980) 1974 год - первое обстоятельное исследование этиологии выраженной умственной отсталости показавшее, что синдромальные формы врожденных пороков развития в структуре олигофрении составляют 29%, а метаболические "синдромы" - 6, 5% (Kaveggia et al. , 1974) 1977 год - первый клинический атлас хромосомных заболеваний человека французских исследователей Jean de Grouchy and Catherine Turleau (de Grouchy, Turleau, 1977). Прекрасно иллюстрованное руководство по синдромологии хромосомных заболеваний 1979 год - обширная публикация John M. Opitz - одного из ведущих американских генетиков, об основных понятиях синдромологии врожденных пороков развития (Opitz JM. , 1979) 1980 год - начало работ по созданию компьтерных баз данных по синдромологии. Одной из самых первых программ по синдромологии появилась австралийская система POSSUM 1987 год - публикация первого на русском языке иллюстрированного словаря по наследственным синдромам (Козлова С. И. , Семанова Е. , Демикова Н. С. , Блинникова О. Е. , 1987). 1992 год - начало выпуска специализированного журнала "Clinical Dysmorphology" (редакторы: Robert Winter, Dian Donnai, Michael Baraitser), в котором публикуются работы по всем аспектам синдромологии врожденных дефектов развития. 1993 год - публикация диагностического обзора множественных врожденных аномалий Robin M. Winter and Michel Baraitser, где кратко систематизированы более 400 "новых" синдромов описанных за последнии годы (Winter, Baraitser, 1993). В дальнейшем эта база данных стала известна как "Лондонская база данных по дисморфологии" (Winter, Baraitser, 1993, 1996).
 Критические периоды в развитии эмбриона и плода
Критические периоды в развитии эмбриона и плода
 Критические периоды в развитии эмбриона и плода 1. От 0 до 10 дней – нет связи с материнским организмом, эмбрион или погибает или развивается. 2. От 10 дней до 12 недель происходит формирование органов и систем, характерно возникновение множественных пороков развития. Значение имеет не столько срок гестации, но и длительность воздействия неблагоприятного фактора. Особенно критичны 3 -4 недели – начало формирования плаценты и хориона. Нарушение ее развития приводит к плацентарной недостаточности, также к гибели эмбриона или развитию гипотрофии плода. 3. 12 -16 недель - формируются наружные половые органы. Введение эстрогенов может привести к дисплазии эпителия матки и влагалища во взрослом состоянии. 4. 18 -22 недели - завершение формирования нервной системы.
Критические периоды в развитии эмбриона и плода 1. От 0 до 10 дней – нет связи с материнским организмом, эмбрион или погибает или развивается. 2. От 10 дней до 12 недель происходит формирование органов и систем, характерно возникновение множественных пороков развития. Значение имеет не столько срок гестации, но и длительность воздействия неблагоприятного фактора. Особенно критичны 3 -4 недели – начало формирования плаценты и хориона. Нарушение ее развития приводит к плацентарной недостаточности, также к гибели эмбриона или развитию гипотрофии плода. 3. 12 -16 недель - формируются наружные половые органы. Введение эстрогенов может привести к дисплазии эпителия матки и влагалища во взрослом состоянии. 4. 18 -22 недели - завершение формирования нервной системы.
 Писал в 1932 г. писал по поводу болезни") Petrus Johannes Waardenburg (1886 – 1979) Писал в 1932 г. писал по поводу болезни Дауна: «Наличие целой группы симптомов у таких больных особенно привлекает внимание. Я хотел бы предложить цитологам проверить, не встречаемся ли мы здесь с примером определенной хромосомной аберрации у человека. Почему бы ей не возникать иногда у человека и почему бы ей не быть, если она не детальна, причиной глубокой аномалии конституции. Нужно ответить на вопрос, не лежит ли в основе монголизма «хромосомная нехватка» или «нерасхождение» , или, наоборот, «хромосомная дупликация» . Моя гипотеза имеет по крайней мере то преимущество, что ее можно проверить. Если она верна, становится понятным и влияние возраста матери. . . »
Petrus Johannes Waardenburg (1886 – 1979) Писал в 1932 г. писал по поводу болезни Дауна: «Наличие целой группы симптомов у таких больных особенно привлекает внимание. Я хотел бы предложить цитологам проверить, не встречаемся ли мы здесь с примером определенной хромосомной аберрации у человека. Почему бы ей не возникать иногда у человека и почему бы ей не быть, если она не детальна, причиной глубокой аномалии конституции. Нужно ответить на вопрос, не лежит ли в основе монголизма «хромосомная нехватка» или «нерасхождение» , или, наоборот, «хромосомная дупликация» . Моя гипотеза имеет по крайней мере то преимущество, что ее можно проверить. Если она верна, становится понятным и влияние возраста матери. . . »
 Терминология Ø Порок развития – морфологический дефект органа, части органа или части тела в результате нарушения внутриутробного развития Ø Дисплазия – результат ненормальной организации клеток в ткани, когда возникшее структурное изменение связано с неправильным развитием отдельного типа ткани Ø Деформации – ненормальные по форме, очертанию или положению части тела, вызванные механическими воздействиями Ø Дизрупции – морфологический дефект органа, части органа или части тела от нарушений кровоснабжения, асфиксии, инфекции или же действия механических сил Аномалии морфогенеза 1. Неполный морфогенез (отсутствие органа или ткани, незаращение, распаж структуры) 2. Чрезмерный морфогенез (дупликация структур, например, полидактилия) 3. Аберрантный морфогенез (эктопическая локализация при отсутствии или нарушении миграции клеток в эмбриогенезе)
Терминология Ø Порок развития – морфологический дефект органа, части органа или части тела в результате нарушения внутриутробного развития Ø Дисплазия – результат ненормальной организации клеток в ткани, когда возникшее структурное изменение связано с неправильным развитием отдельного типа ткани Ø Деформации – ненормальные по форме, очертанию или положению части тела, вызванные механическими воздействиями Ø Дизрупции – морфологический дефект органа, части органа или части тела от нарушений кровоснабжения, асфиксии, инфекции или же действия механических сил Аномалии морфогенеза 1. Неполный морфогенез (отсутствие органа или ткани, незаращение, распаж структуры) 2. Чрезмерный морфогенез (дупликация структур, например, полидактилия) 3. Аберрантный морфогенез (эктопическая локализация при отсутствии или нарушении миграции клеток в эмбриогенезе)
 Терминология при обнаружении морфологических дефектов Ø Гипоплазия или гиперплазия – недоразвитие или избыточное развитие организма, органа или ткани в результате снижения или увеличесния числа клеток Ø Гипотрофия или гипертрофия – уменьшение или увеличение в размере клеток, тканей и органов Ø Агенезия – отсутствие части тела в результате повреждения закладки для последующего развития Ø Атрофия – снижение нормального развития массы ткани (тканей) из-за снижения размера и/или количества клеток
Терминология при обнаружении морфологических дефектов Ø Гипоплазия или гиперплазия – недоразвитие или избыточное развитие организма, органа или ткани в результате снижения или увеличесния числа клеток Ø Гипотрофия или гипертрофия – уменьшение или увеличение в размере клеток, тканей и органов Ø Агенезия – отсутствие части тела в результате повреждения закладки для последующего развития Ø Атрофия – снижение нормального развития массы ткани (тканей) из-за снижения размера и/или количества клеток

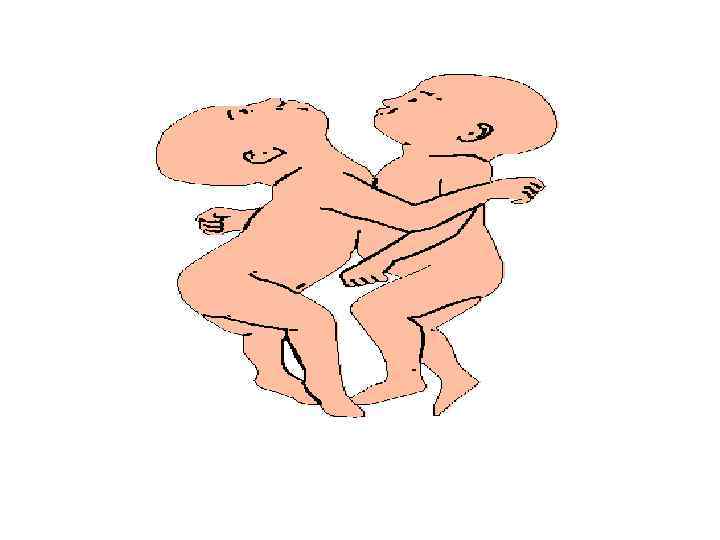
 При монохориальном моноамниотическом типе двоен Вероятность 1: 200 000 родов Симские близнецы Энг и Чанг (1911 -1973)
При монохориальном моноамниотическом типе двоен Вероятность 1: 200 000 родов Симские близнецы Энг и Чанг (1911 -1973)
 пигопаги торакопаги краниопаги
пигопаги торакопаги краниопаги
 – 35 -40% случаев, срастание в области грудной") Типы сросшихся близнецов 1. Торокопаги (thorocopagus) – 35 -40% случаев, срастание в области грудной клетки, в патологический процесс вовлечено сердце 2. Омфалопаги (omphalopagus) – 34% случаев, срастание в нижней части грудной клетки, сердце не затронуто, но общие печень, пищеварительный тракт, диафрагма 3. Ксифопаги (xiphopagus) – срастание хрящей грудной клетки 4. Илиопаги (iliopagus) – 19% случаев, срастание в подвздошных областях, спиной к спине, включая ягодицы 5. Цефалопаги (cephalopagus) – срастание головами и туловищами, обычно не жизнеспособны 6. Краниопаги (craniopagus) – 2% случаев, срастание черепами при раздельных туловищах 7. Паразитарные краниопаги – две сросшиеся головы с одним туловищем 8. Дицефалы (dicephalus) – две головы, одно туловище и две, три или четыре руки (дицефал дибрахиус, трибрахиус или тетрабрахиус) 9. Ишиопаги (ischiopagus) – 6% случаев, соединение нижних частей тела, объединение таза, крестца, могут быть сросшиеся позвоночники 10. Ишио-омфалопаги (ischio-omphalopagus) – срастание позвоночниками в форме У, 4 руки и 2 или 3 ноги, общие репродуктивная и выделительная системы 11. Парапаги (parapagus) – 5% случаев, срастание боками, иногда вовлечено сердце 12. Дипросопус (diprosopus) – одна голова с двумя лицами, расположенными бок о бок 13. Пигопаги (pigopagus) – срастание в области крестца 14. Трицефалы – двойное или тройное сращение, у трех близнецов может быть один торс и три головы
Типы сросшихся близнецов 1. Торокопаги (thorocopagus) – 35 -40% случаев, срастание в области грудной клетки, в патологический процесс вовлечено сердце 2. Омфалопаги (omphalopagus) – 34% случаев, срастание в нижней части грудной клетки, сердце не затронуто, но общие печень, пищеварительный тракт, диафрагма 3. Ксифопаги (xiphopagus) – срастание хрящей грудной клетки 4. Илиопаги (iliopagus) – 19% случаев, срастание в подвздошных областях, спиной к спине, включая ягодицы 5. Цефалопаги (cephalopagus) – срастание головами и туловищами, обычно не жизнеспособны 6. Краниопаги (craniopagus) – 2% случаев, срастание черепами при раздельных туловищах 7. Паразитарные краниопаги – две сросшиеся головы с одним туловищем 8. Дицефалы (dicephalus) – две головы, одно туловище и две, три или четыре руки (дицефал дибрахиус, трибрахиус или тетрабрахиус) 9. Ишиопаги (ischiopagus) – 6% случаев, соединение нижних частей тела, объединение таза, крестца, могут быть сросшиеся позвоночники 10. Ишио-омфалопаги (ischio-omphalopagus) – срастание позвоночниками в форме У, 4 руки и 2 или 3 ноги, общие репродуктивная и выделительная системы 11. Парапаги (parapagus) – 5% случаев, срастание боками, иногда вовлечено сердце 12. Дипросопус (diprosopus) – одна голова с двумя лицами, расположенными бок о бок 13. Пигопаги (pigopagus) – срастание в области крестца 14. Трицефалы – двойное или тройное сращение, у трех близнецов может быть один торс и три головы

 Врожденные пороки развития ØГаметопатии – патология, связанная с изменениями наследственного материала в процессе закладки и развития половых клеток родителей (гаметогенез), либо во время оплодотворения и в период дробления. Исходы: гибель зародыша, самопроизвольный аборт, мертворождение, грубые пороки развития, наследственные заболевания, в т. ч. Хромосомным заболеваниям и ферментопатиям ØБластопатии – патология в период 4 -15 дни после оплодотворения. Исходы: эктопическая имплантация, нарушения формирования плаценты, часто самопроизвольные аборты, грубые уродства, напр. циклопия, сирингомиелия ØЭмбриопатии – патология с 16 -20 дня после оплодотворения до конца 8 -ой недели внутриутробного развития Исходы: ВПР, гибель эмбриона, самопроизвольный выкидыш, преждевременные роды ØФетопатии – с 9 -ой недели развития до родов. Ранние фетопатии (9 -29 недели беременности) грубые дефекты ЦНС (микроцефалия, микрогерия и др). Поздние фетопатии (с 29 -ой недели) – функциональные расстройства систем и органов
Врожденные пороки развития ØГаметопатии – патология, связанная с изменениями наследственного материала в процессе закладки и развития половых клеток родителей (гаметогенез), либо во время оплодотворения и в период дробления. Исходы: гибель зародыша, самопроизвольный аборт, мертворождение, грубые пороки развития, наследственные заболевания, в т. ч. Хромосомным заболеваниям и ферментопатиям ØБластопатии – патология в период 4 -15 дни после оплодотворения. Исходы: эктопическая имплантация, нарушения формирования плаценты, часто самопроизвольные аборты, грубые уродства, напр. циклопия, сирингомиелия ØЭмбриопатии – патология с 16 -20 дня после оплодотворения до конца 8 -ой недели внутриутробного развития Исходы: ВПР, гибель эмбриона, самопроизвольный выкидыш, преждевременные роды ØФетопатии – с 9 -ой недели развития до родов. Ранние фетопатии (9 -29 недели беременности) грубые дефекты ЦНС (микроцефалия, микрогерия и др). Поздние фетопатии (с 29 -ой недели) – функциональные расстройства систем и органов
 Репродуктивные потери на разных сроках беременности В среднем 14 -22% Число выживших новорожденных с врожденными пороками развития (ВПР) не более 6% ØПервые 2 недели после зачатия: до 75% (нарушения оплодотворения и имплантации) Ø 3 -6 недели беременности: не менее 10% (резорбции и микроаборты) Доля хромосомных нарушений от общего числа нарушений: Ø 50% на 3 -6 неделях беременности Ø 41 -50% на 8 -11 неделях беременности Ø 30% к 16 -19 неделям беременности
Репродуктивные потери на разных сроках беременности В среднем 14 -22% Число выживших новорожденных с врожденными пороками развития (ВПР) не более 6% ØПервые 2 недели после зачатия: до 75% (нарушения оплодотворения и имплантации) Ø 3 -6 недели беременности: не менее 10% (резорбции и микроаборты) Доля хромосомных нарушений от общего числа нарушений: Ø 50% на 3 -6 неделях беременности Ø 41 -50% на 8 -11 неделях беременности Ø 30% к 16 -19 неделям беременности
 Вредные воздействия на ранних этапах эмбрионального развития приводят к грубым нарушениям морфогенеза - эмбриопатиям Конюхов, 1980
Вредные воздействия на ранних этапах эмбрионального развития приводят к грубым нарушениям морфогенеза - эмбриопатиям Конюхов, 1980
 Возможные исходы беременностей и родов при воздействии вредных факторов на разных этапах эмбриогенеза
Возможные исходы беременностей и родов при воздействии вредных факторов на разных этапах эмбриогенеза
 Факторы риска развития нарушений у зародыша ØВозраст матери ØБолезнь и лечение в период беременности ØПрофессиональная вредность Тератология изучает аномальное развитие плода и стремится выяснить причины и механизмы патологического развития. В качестве тератогена может выступать вещество или физический агент, нарушающий структурное или функциональное развитие Примерно 1 из 400 живорожденных детей имеет уродства, обусловленные тератогенными веществами. Тератогены могут быть причиной 10% всех врожденных дефектов.
Факторы риска развития нарушений у зародыша ØВозраст матери ØБолезнь и лечение в период беременности ØПрофессиональная вредность Тератология изучает аномальное развитие плода и стремится выяснить причины и механизмы патологического развития. В качестве тератогена может выступать вещество или физический агент, нарушающий структурное или функциональное развитие Примерно 1 из 400 живорожденных детей имеет уродства, обусловленные тератогенными веществами. Тератогены могут быть причиной 10% всех врожденных дефектов.
 Факторы, участвующие в тератогенезе человека ØМедикаментозные средства, используемые вовремя беременности: талидомид, андрогенные гормоны, тетрациклин, аминоптерин, изотретион и др. ØМетаболические дисфункции у беременных женщин: сахарный диабет, недостаточность йода, голодание ØФизические факторы окружающей среды: иррадиация, полихлоринбифенилы, метилмеркурий. ØВещества добровольно принимаемые или вдыхаемые внутрь: курение сигарет, алкоголь, наркотики. ØМатеринские инфекции: сифилис, краснуха, цитомегалия, токсоплазмоз, вирус иммунодефицита человека.
Факторы, участвующие в тератогенезе человека ØМедикаментозные средства, используемые вовремя беременности: талидомид, андрогенные гормоны, тетрациклин, аминоптерин, изотретион и др. ØМетаболические дисфункции у беременных женщин: сахарный диабет, недостаточность йода, голодание ØФизические факторы окружающей среды: иррадиация, полихлоринбифенилы, метилмеркурий. ØВещества добровольно принимаемые или вдыхаемые внутрь: курение сигарет, алкоголь, наркотики. ØМатеринские инфекции: сифилис, краснуха, цитомегалия, токсоплазмоз, вирус иммунодефицита человека.
 Шесть принципов тератологии ØЧувствительность к тератогену зависит от генотипа и взаимодействия генотипа с факторами окружающей среды ØСтепень чувствительности к тератогену зависит от стадии внутриутробного развития ØТератогенные агенты имеют специфические механизмы действия на клетки и ткани ØФинал ненормального развития: смерть, уродства, задержка роста или функциональные расстройства. Степень выраженности зависит от стадии эмбриогенеза ØТератогенный эффект на развивающуюся ткань зависит от природы агента ØСтепень тяжести аномалии развития возрастает с увеличением дозы тератогена
Шесть принципов тератологии ØЧувствительность к тератогену зависит от генотипа и взаимодействия генотипа с факторами окружающей среды ØСтепень чувствительности к тератогену зависит от стадии внутриутробного развития ØТератогенные агенты имеют специфические механизмы действия на клетки и ткани ØФинал ненормального развития: смерть, уродства, задержка роста или функциональные расстройства. Степень выраженности зависит от стадии эмбриогенеза ØТератогенный эффект на развивающуюся ткань зависит от природы агента ØСтепень тяжести аномалии развития возрастает с увеличением дозы тератогена
 Фетальный алкогольный синдром
Фетальный алкогольный синдром
 Фетальный алкогольный синдром Синонимы: алкогольный синдром плода, алкогольная эмбриофетопатия, эмбриональный алкогольный синдром, фетальные алкогольные эффекты — различные как по сочетанию, так и по степени выраженности отклонения в психофизическом развитии ребёнка, причиной которых является употребление женщиной алкоголя до и во время беременности 1) Мозговые аномалии и расстройства, связанные с деятельностью центральной нервной системы, включая неврологические аномалии, умственную отсталость, нарушения поведения, нарушения интеллекта иили аномалии структуры мозга; 2) Пренатальный и/или постнатальный дефицит роста и веса. 3) Специфические особенности строения лица: короткая глазная щель сглаженный губной желобок, тонкая верхняя губа (кайма верхней губы) Дети с ФАС: • отстают в росте и весе • имеют характерные особенности лица — лицевые аномалии • могут иметь проблемы со слухом и зрением • хуже обучаются элементарным вещам • имеют проблемы с памятью и вниманием и трудности в обучении в школе • хуже контролируют свои эмоции и свое поведение • могут нуждаться в специальных педагогах и обучении в специальных школах • часто недостаточно осознают последствия своих поступков • могут совершать асоциальные поступки и вступать в конфликт с законом • всю жизнь нуждаются в социальной защите и медицинском сопровождении
Фетальный алкогольный синдром Синонимы: алкогольный синдром плода, алкогольная эмбриофетопатия, эмбриональный алкогольный синдром, фетальные алкогольные эффекты — различные как по сочетанию, так и по степени выраженности отклонения в психофизическом развитии ребёнка, причиной которых является употребление женщиной алкоголя до и во время беременности 1) Мозговые аномалии и расстройства, связанные с деятельностью центральной нервной системы, включая неврологические аномалии, умственную отсталость, нарушения поведения, нарушения интеллекта иили аномалии структуры мозга; 2) Пренатальный и/или постнатальный дефицит роста и веса. 3) Специфические особенности строения лица: короткая глазная щель сглаженный губной желобок, тонкая верхняя губа (кайма верхней губы) Дети с ФАС: • отстают в росте и весе • имеют характерные особенности лица — лицевые аномалии • могут иметь проблемы со слухом и зрением • хуже обучаются элементарным вещам • имеют проблемы с памятью и вниманием и трудности в обучении в школе • хуже контролируют свои эмоции и свое поведение • могут нуждаться в специальных педагогах и обучении в специальных школах • часто недостаточно осознают последствия своих поступков • могут совершать асоциальные поступки и вступать в конфликт с законом • всю жизнь нуждаются в социальной защите и медицинском сопровождении
 Ионизирующая радиация ØПри облучении в срок менее 2 недель – «все или ничего» . ØПри воздействии на 2 -18 неделях гестации возможны задержки внутриутробного развития и последующая умственная отсталость, сопровождающаяся микроцефалией, микроофтальмией и катарактой. ØПри облучении беременной после 18 недель гестации у плода может возникнуть состояние, похожее на радиационную болезнь взрослых (потеря волос, гипоплазия костного мозга, кожные изменения и др).
Ионизирующая радиация ØПри облучении в срок менее 2 недель – «все или ничего» . ØПри воздействии на 2 -18 неделях гестации возможны задержки внутриутробного развития и последующая умственная отсталость, сопровождающаяся микроцефалией, микроофтальмией и катарактой. ØПри облучении беременной после 18 недель гестации у плода может возникнуть состояние, похожее на радиационную болезнь взрослых (потеря волос, гипоплазия костного мозга, кожные изменения и др).
 Лекарственные вещества, обладающие тератогенным эффектом Агент Механизм действия Наиболее частые врожденные пороки Пренатальная диагностика Лекарства, алкоголь Повышенная смерть клеток Фетальный алкогольный синдром; задержка развития; отклонения ЦНС; характерное выражение лица УЗИ для диагностики аномалий Аминоптерин и Нарушения антифолаты деления клеток Задержка развития; дефекты скелета; пороки развития ЦНС (анэнцефалия и др). УЗИ для диагностики аномалий Кокаин вазоконстрикция Задержка развития плода; недоношенность; микроцефалия; церебральный инфаркт; нейроповеденческие расстройства Наблюдение группы высокого риска Изотретиноин Гибель клеток Краниофациальные пороки; дефекты нервной трубки; сердечно-сосудистые дефекты УЗИ Карбонат лития - Дефекты сердца; дефекты нервной трубки Эхокардиографи я плода Талидомид Нарушения деления клеток Ненормальное развитие конечностей (миромелия, амелия) УЗИ Полихлоратные бифенилы Внутриутробная задержка развития; нарушение окраски кожи
Лекарственные вещества, обладающие тератогенным эффектом Агент Механизм действия Наиболее частые врожденные пороки Пренатальная диагностика Лекарства, алкоголь Повышенная смерть клеток Фетальный алкогольный синдром; задержка развития; отклонения ЦНС; характерное выражение лица УЗИ для диагностики аномалий Аминоптерин и Нарушения антифолаты деления клеток Задержка развития; дефекты скелета; пороки развития ЦНС (анэнцефалия и др). УЗИ для диагностики аномалий Кокаин вазоконстрикция Задержка развития плода; недоношенность; микроцефалия; церебральный инфаркт; нейроповеденческие расстройства Наблюдение группы высокого риска Изотретиноин Гибель клеток Краниофациальные пороки; дефекты нервной трубки; сердечно-сосудистые дефекты УЗИ Карбонат лития - Дефекты сердца; дефекты нервной трубки Эхокардиографи я плода Талидомид Нарушения деления клеток Ненормальное развитие конечностей (миромелия, амелия) УЗИ Полихлоратные бифенилы Внутриутробная задержка развития; нарушение окраски кожи
 Лекарственные вещества, обладающие тератогенным эффектом Агент Механизм действия Наиболее частые врожденные пороки Пренатальная диагностика Метотерксан Гибель клеток Множественные аномалии, особенно скелетные (лицо, череп, конечности, позвоночник); гидроцефалия; расщепление верхнего неба УЗИ Фенитоин (дилантин) Гибель клеток Фетальные шидонтаиновый синдром; задержка развития; микроцефалия; умственная отсталость; расщепление губы, твердого неба УЗИ Вальпроиковая кислота, Вальфарин Нарушение метаболизма кальция и витамина К Фетальные вальфариновый УЗИ синдром: гипоплазия носа, точечные эпифизы, аномалии глаз, умственная отсталость Метиловая ртуть Болезнь Минимата: церебральные параличи; микроцефалия; умственная отсталость; слепота
Лекарственные вещества, обладающие тератогенным эффектом Агент Механизм действия Наиболее частые врожденные пороки Пренатальная диагностика Метотерксан Гибель клеток Множественные аномалии, особенно скелетные (лицо, череп, конечности, позвоночник); гидроцефалия; расщепление верхнего неба УЗИ Фенитоин (дилантин) Гибель клеток Фетальные шидонтаиновый синдром; задержка развития; микроцефалия; умственная отсталость; расщепление губы, твердого неба УЗИ Вальпроиковая кислота, Вальфарин Нарушение метаболизма кальция и витамина К Фетальные вальфариновый УЗИ синдром: гипоплазия носа, точечные эпифизы, аномалии глаз, умственная отсталость Метиловая ртуть Болезнь Минимата: церебральные параличи; микроцефалия; умственная отсталость; слепота
 Инфекционные агенты, обладающие тератогенным эффектом Агент Механизм действия Наиболее частые врожденные пороки Пренатальная диагностика Цитомегаловирус Микроцефалия; гидроцефалия; церебральные параличи; хориоретинит, потеря чувствительности нервами; психомоторная/умственная задержка УЗИ Вирус простого герпеса Хориоретинит; гидранэнцефалия Вирус иммунодефицита человека Нарушение роста; микроцефалия; выступающий лоб; плоская переносица; гипертелоризм Вирус краснухи Задержка развития; отклонения в развитии сердца; дефекты глаз; потеря слуха Токсоплазма Гонди Микроцефалия; умственная задержка Трепонема паллидум Гидроцефалия; врожденная глухота; умственная задержка УЗИ Вирус ветряной оспы Гидроцефалия; парезы конечностей; судороги; пороки развития глаз; умственная отсталость УЗИ
Инфекционные агенты, обладающие тератогенным эффектом Агент Механизм действия Наиболее частые врожденные пороки Пренатальная диагностика Цитомегаловирус Микроцефалия; гидроцефалия; церебральные параличи; хориоретинит, потеря чувствительности нервами; психомоторная/умственная задержка УЗИ Вирус простого герпеса Хориоретинит; гидранэнцефалия Вирус иммунодефицита человека Нарушение роста; микроцефалия; выступающий лоб; плоская переносица; гипертелоризм Вирус краснухи Задержка развития; отклонения в развитии сердца; дефекты глаз; потеря слуха Токсоплазма Гонди Микроцефалия; умственная задержка Трепонема паллидум Гидроцефалия; врожденная глухота; умственная задержка УЗИ Вирус ветряной оспы Гидроцефалия; парезы конечностей; судороги; пороки развития глаз; умственная отсталость УЗИ
 Календари действия тератогенов Ø 23 -28 -й дни – анэнцефалия или энцефалоцеле Ø 30 -42 -й дни – аномалии базальных ганглиев Ø 5 -й месяц внутриутробной жизни – патология мозолистого тела талидомид Ø 35 -й день гестации – пороки развития ушной раковины Ø 43 -45 -й дни – пороки развития конечностей (фокомелия) Памятник жертвам талидомида
Календари действия тератогенов Ø 23 -28 -й дни – анэнцефалия или энцефалоцеле Ø 30 -42 -й дни – аномалии базальных ганглиев Ø 5 -й месяц внутриутробной жизни – патология мозолистого тела талидомид Ø 35 -й день гестации – пороки развития ушной раковины Ø 43 -45 -й дни – пороки развития конечностей (фокомелия) Памятник жертвам талидомида
 ØКраниофациальные: сросшиеся брови, седловидный нос, плоский затылок, загнутый нос, короткая") Малые аномалии развития (МАР) ØКраниофациальные: сросшиеся брови, седловидный нос, плоский затылок, загнутый нос, короткая шея, плоский профиль лица, затылочная шпора (выступающая затылочная кость), широкая и уплощенная переносица, скошенный лоб ØГлазные: эпикант, короткие глазные щели, синие склеры, узкие глазные щели, гипотелоризм, гипертелоризм, коломба век и радужки, птоз, гетерохромия радужки, двойной ряд ресниц и др. ØАномалии ротовой полости: расщелина язычка, скошенный подбородок, толстые губы с бороздами, микрогнатия, высокое небо, прогения, микрогения, прогнатия ØУшные аномалии: низкое расположение, асимметрия длины, приросшая мочка, большие уши, недоразвитие ушного завитка, отсутствие козелка, оттопыренные уши, околоушные придатки, наршения формы наружного уха ØАномалии рук: короткий мизинец, гипоплазия ногтей, клинодактилия, синдактилия, полидактилия, уплощение ногтевых фаланг, поперечная складка на ладони, брахидактилия, арахнодактилия ØАномалии ног: синдактилия, полидактилия, широкий промежуток между 1 -м и 2 -м пальцами, сандалевидная щель, широкий большой короткий палец ØАномалии кожи и туловища: отсутствие или гипоплазия сосков, гемангиомы, пигментация, соски на разном уровне, гирсутизм, избыток кожи, низкий рост волос
Малые аномалии развития (МАР) ØКраниофациальные: сросшиеся брови, седловидный нос, плоский затылок, загнутый нос, короткая шея, плоский профиль лица, затылочная шпора (выступающая затылочная кость), широкая и уплощенная переносица, скошенный лоб ØГлазные: эпикант, короткие глазные щели, синие склеры, узкие глазные щели, гипотелоризм, гипертелоризм, коломба век и радужки, птоз, гетерохромия радужки, двойной ряд ресниц и др. ØАномалии ротовой полости: расщелина язычка, скошенный подбородок, толстые губы с бороздами, микрогнатия, высокое небо, прогения, микрогения, прогнатия ØУшные аномалии: низкое расположение, асимметрия длины, приросшая мочка, большие уши, недоразвитие ушного завитка, отсутствие козелка, оттопыренные уши, околоушные придатки, наршения формы наружного уха ØАномалии рук: короткий мизинец, гипоплазия ногтей, клинодактилия, синдактилия, полидактилия, уплощение ногтевых фаланг, поперечная складка на ладони, брахидактилия, арахнодактилия ØАномалии ног: синдактилия, полидактилия, широкий промежуток между 1 -м и 2 -м пальцами, сандалевидная щель, широкий большой короткий палец ØАномалии кожи и туловища: отсутствие или гипоплазия сосков, гемангиомы, пигментация, соски на разном уровне, гирсутизм, избыток кожи, низкий рост волос
 Патологии неонатального периода Øнедоношенность Øасфиксия Øврожденные пороки развития (4, 3 – 55 на 1000 новорожденных) Частота врожденных пороков развития в разных выборках Исследуемая группа Спонтанные аборты: 1 триместр 2 триместр Новорожденные и дети до 1 года жизни Плоды, умершие в перинатальном периоде Дети, умершие: до 1 года жизни от 1 года до 9 лет от 10 лет до 14 лет Частота, % 80 -85 25 2 -5 25 -30 25 20 7, 5
Патологии неонатального периода Øнедоношенность Øасфиксия Øврожденные пороки развития (4, 3 – 55 на 1000 новорожденных) Частота врожденных пороков развития в разных выборках Исследуемая группа Спонтанные аборты: 1 триместр 2 триместр Новорожденные и дети до 1 года жизни Плоды, умершие в перинатальном периоде Дети, умершие: до 1 года жизни от 1 года до 9 лет от 10 лет до 14 лет Частота, % 80 -85 25 2 -5 25 -30 25 20 7, 5
 Сроки возникновения врожденных уродств Локализация порока развития лицо Порок развития Время возникновения 8 -12 недель Атрезия пищевода +трахеоэзофагальная фистула 4 неделя Атрезия прямой кишки (с фистулой) 6 неделя Незавершенный поворот кишечника (мальротация) 10 неделя Омфалоцеле (грыжа пупочного канатика) 10 неделя Атрезия поджелудочной железы Центральная нервная системы 5 -7 недель Расщепление заднего неба Желудочнокишечный тракт Расщепление губы 7 -8 недели Анэнцефалия 3 -4 недели Менингомиелоцеле 4 неделя Сердечно. Транспозиция больших сосудов сосудистая система Дефект желудочковых перегородок 5 неделя 5 -6 недели
Сроки возникновения врожденных уродств Локализация порока развития лицо Порок развития Время возникновения 8 -12 недель Атрезия пищевода +трахеоэзофагальная фистула 4 неделя Атрезия прямой кишки (с фистулой) 6 неделя Незавершенный поворот кишечника (мальротация) 10 неделя Омфалоцеле (грыжа пупочного канатика) 10 неделя Атрезия поджелудочной железы Центральная нервная системы 5 -7 недель Расщепление заднего неба Желудочнокишечный тракт Расщепление губы 7 -8 недели Анэнцефалия 3 -4 недели Менингомиелоцеле 4 неделя Сердечно. Транспозиция больших сосудов сосудистая система Дефект желудочковых перегородок 5 неделя 5 -6 недели
 Число детей-инвалидов с соматическими и психическими расстройствами 1996 год – 462 275 детей 2000 год – 554 867 детей 2002 год – 620 342 ребенка Снижение перинатальной смертности за последние 30 -35 лет Австрия с 28, 5 до 5% Финляндия с 27, 5 до 5, 1% Япония с 37, 8 до 7% в 5, 7 раз в 5, 4 раза
Число детей-инвалидов с соматическими и психическими расстройствами 1996 год – 462 275 детей 2000 год – 554 867 детей 2002 год – 620 342 ребенка Снижение перинатальной смертности за последние 30 -35 лет Австрия с 28, 5 до 5% Финляндия с 27, 5 до 5, 1% Япония с 37, 8 до 7% в 5, 7 раз в 5, 4 раза
 Снижение перинатальной и ранней неонатальной смертности и увеличение ВПР среди умерших и живых новорожденных
Снижение перинатальной и ранней неонатальной смертности и увеличение ВПР среди умерших и живых новорожденных
 Примеры ассоциированных пороков развития Уродства/диагноз Возможные ассоциации Аниридия Опухоль Вильмса Единственный резец зуба в центре Дефект по средней линии в гоовном мозге Гетерохромия радужки Глухота Радиальная аплазия Трахеоэзофагиальный свищ, аноректальный порок, врожденный порок сердца Di George последовательность Иммунный дефицит, гипокальцемия, врожденный порок сердца, расщепление неба Синдром Дауна Врожденный порок сердца, атрезия 12 перстной кишки, болезнь Гиршпрунга Синдром Тернера Коарктация аорты
Примеры ассоциированных пороков развития Уродства/диагноз Возможные ассоциации Аниридия Опухоль Вильмса Единственный резец зуба в центре Дефект по средней линии в гоовном мозге Гетерохромия радужки Глухота Радиальная аплазия Трахеоэзофагиальный свищ, аноректальный порок, врожденный порок сердца Di George последовательность Иммунный дефицит, гипокальцемия, врожденный порок сердца, расщепление неба Синдром Дауна Врожденный порок сердца, атрезия 12 перстной кишки, болезнь Гиршпрунга Синдром Тернера Коарктация аорты
, из них 15 924 – аутосомные") Описано 16 996 наследственных фенотипов (к 2006 г), из них 15 924 – аутосомные (отсеквенированы 10 361) 953 – Х-сцепленные (отсеквенированы 483) 56 – У-сцеплены (отсеквенированы 48) 63 – митохондриальных (отсеквенированы 37) Число наследственных болезней – более 4, 5 тысяч 1700 аутосомно-доминантные 1700 аутосомно-рецессивные 300 Х-сцепленные 4 У-сцепленные 22 с митохондриальным типом наследования
Описано 16 996 наследственных фенотипов (к 2006 г), из них 15 924 – аутосомные (отсеквенированы 10 361) 953 – Х-сцепленные (отсеквенированы 483) 56 – У-сцеплены (отсеквенированы 48) 63 – митохондриальных (отсеквенированы 37) Число наследственных болезней – более 4, 5 тысяч 1700 аутосомно-доминантные 1700 аутосомно-рецессивные 300 Х-сцепленные 4 У-сцепленные 22 с митохондриальным типом наследования
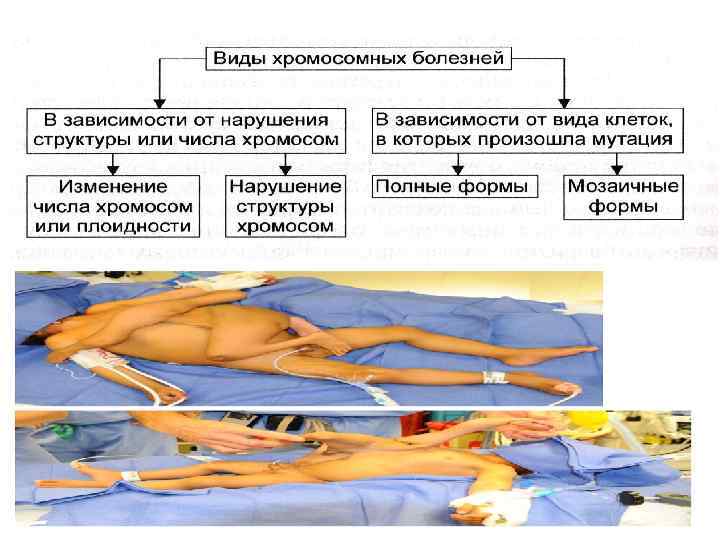
 Врожденные наследственные заболевания болезни, обусловленные нарушениями в процессах хранения, передачи и реализации генетической информации Качественно-количественная классификация: ØИзменение числа хромосом – хромосомные болезни: уменьшение или увеличение числа хромосом. ØКачественное изменение ДНК – генные болезни, мутации ядерной ДНК – мутации митохондриальной ДНК (Патологические нарушения клеточного энергетического обмена могут проявляться в виде дефектов различных звеньев в цикле Кребса, в дыхательной цепи, процессах бета-окисления и т. д) Дифференциация наследственных заболеваний: ØМоногенные – аутосомно-доминантные – аутосомно-рецессивные – сцепленные с полом ØПолигенные (рак, сахарный диабет, шизофрения, эпилепсия, ИБС, гипертензия и др. ) ØХромосомные аберрации (синдромы Дауна, Клайнфельтера, Шершевского. Тернера, Эдвардса, «кошачьего крика» и др. )
Врожденные наследственные заболевания болезни, обусловленные нарушениями в процессах хранения, передачи и реализации генетической информации Качественно-количественная классификация: ØИзменение числа хромосом – хромосомные болезни: уменьшение или увеличение числа хромосом. ØКачественное изменение ДНК – генные болезни, мутации ядерной ДНК – мутации митохондриальной ДНК (Патологические нарушения клеточного энергетического обмена могут проявляться в виде дефектов различных звеньев в цикле Кребса, в дыхательной цепи, процессах бета-окисления и т. д) Дифференциация наследственных заболеваний: ØМоногенные – аутосомно-доминантные – аутосомно-рецессивные – сцепленные с полом ØПолигенные (рак, сахарный диабет, шизофрения, эпилепсия, ИБС, гипертензия и др. ) ØХромосомные аберрации (синдромы Дауна, Клайнфельтера, Шершевского. Тернера, Эдвардса, «кошачьего крика» и др. )
 Классификация хромосомных аномалий 1. Геномные 2. Структурные 3. Генные мутации
Классификация хромосомных аномалий 1. Геномные 2. Структурные 3. Генные мутации
 Геномные мутации Гетероплоидия – любое численное изменение количества хромосом • Кратное гаплоидному набору: Триплоидия (3 n=69) Тетраплоидия (4 n=92) • Некратное гаплоидному набору (анеуплоидия) Трисомия (2 n+1=47) Полисомия (2 n+x=46+x) Моносомия (2 n-1=45) Нуллисомия (2 n-2=44)
Геномные мутации Гетероплоидия – любое численное изменение количества хромосом • Кратное гаплоидному набору: Триплоидия (3 n=69) Тетраплоидия (4 n=92) • Некратное гаплоидному набору (анеуплоидия) Трисомия (2 n+1=47) Полисомия (2 n+x=46+x) Моносомия (2 n-1=45) Нуллисомия (2 n-2=44)
 форма мутации – если она присутствует во всех клетках") Геномные мутации • Истинная (полная) форма мутации – если она присутствует во всех клетках зародыша • Мозаичная форма гетероплоидии – мутация выявляется лишь в отдельных клетках • Химеризм – выявление клеток разного генотипа • Однородительская дисомия – одна пара гомологичных хромосом представлена хромосомами только одного из родителей
Геномные мутации • Истинная (полная) форма мутации – если она присутствует во всех клетках зародыша • Мозаичная форма гетероплоидии – мутация выявляется лишь в отдельных клетках • Химеризм – выявление клеток разного генотипа • Однородительская дисомия – одна пара гомологичных хромосом представлена хромосомами только одного из родителей
 Хромосомные мутации Хросомные абберации – сбалансированные или несбалансированные изменения структуры одной или нескольких хромосом • Межхромосомные перестройки (транслокации) – перемещение генетического материала между хромосомами Реципрокные транслокации – сбалансированный обмен фрагментами между двумя и более негомологичными хромосомами Нереципрокные транслокации – несбалансированный обмен фрагментами Робертсоновские транслокации – воссоединение плеч двух акроцентрических хромосом в околоцентромерных районах Инсерция – перемещение фрагмента одной хромосомы внутрь другой
Хромосомные мутации Хросомные абберации – сбалансированные или несбалансированные изменения структуры одной или нескольких хромосом • Межхромосомные перестройки (транслокации) – перемещение генетического материала между хромосомами Реципрокные транслокации – сбалансированный обмен фрагментами между двумя и более негомологичными хромосомами Нереципрокные транслокации – несбалансированный обмен фрагментами Робертсоновские транслокации – воссоединение плеч двух акроцентрических хромосом в околоцентромерных районах Инсерция – перемещение фрагмента одной хромосомы внутрь другой
 Хромосомные мутации • Внутрихросомные перестройки Делеция – утрата части хромосомы Дупликация – удвоение части хромосомы Инверсия – переворот фрагмента хромосомы на 1800, затрагивающий центромеру (перицентрическая инверсия) и не включающий ее (парацентрическая инверсия) Изохромосома – хромосома с генетически идентичными плечами Кольцевые хромосомы – замкнутые кольца с одной или двумя центромерами
Хромосомные мутации • Внутрихросомные перестройки Делеция – утрата части хромосомы Дупликация – удвоение части хромосомы Инверсия – переворот фрагмента хромосомы на 1800, затрагивающий центромеру (перицентрическая инверсия) и не включающий ее (парацентрическая инверсия) Изохромосома – хромосома с генетически идентичными плечами Кольцевые хромосомы – замкнутые кольца с одной или двумя центромерами
 Генные мутации – аномалии участка ДНК размером от одного нуклеотида до гена Делеция – утрата участка гена Дупликация – удвоение сегмента ДНК Инверсия – поворот сегмента ДНК на 1800 Инсерция – вставка фрагментов ДНК Трансверсия – замена пуринового основания на пиримидиновое, или наоборот в одном кодоне Транзикция – замена одного пуринового основания на другое пуриновое основание, или одного пиримидинового на другое пиримидиновое в структуре кодона
Генные мутации – аномалии участка ДНК размером от одного нуклеотида до гена Делеция – утрата участка гена Дупликация – удвоение сегмента ДНК Инверсия – поворот сегмента ДНК на 1800 Инсерция – вставка фрагментов ДНК Трансверсия – замена пуринового основания на пиримидиновое, или наоборот в одном кодоне Транзикция – замена одного пуринового основания на другое пуриновое основание, или одного пиримидинового на другое пиримидиновое в структуре кодона
 – не имеют фенотипического выражения • Миссенс-мутации") Типы генных мутаций • Нейтральные мутации (молчащие) – не имеют фенотипического выражения • Миссенс-мутации – замена нуклеотида в кодирующей части гена приводит к замене аминокислоты в полипептиде • Нонсенс-мутации – замена нуклеотида в кодирующей части гена приводит к образованию стоп-кодона и прекращению трансляции • Регуляторная мутация – в 5`- или 3`-нетранслируемых областях нарушает экспрессию гена • Динамическая мутация – обусловлена увеличением числа тринуклеотидных повторов в функционально значимых частях гена.
Типы генных мутаций • Нейтральные мутации (молчащие) – не имеют фенотипического выражения • Миссенс-мутации – замена нуклеотида в кодирующей части гена приводит к замене аминокислоты в полипептиде • Нонсенс-мутации – замена нуклеотида в кодирующей части гена приводит к образованию стоп-кодона и прекращению трансляции • Регуляторная мутация – в 5`- или 3`-нетранслируемых областях нарушает экспрессию гена • Динамическая мутация – обусловлена увеличением числа тринуклеотидных повторов в функционально значимых частях гена.
 Группы наследственных заболеваний • Гаметические – вызванные вследствие мутаций в половых клетках • Соматические – возникающие вследствие мутаций в соматических клетках • Смешанные – развивающиеся как следствие комбинаций мутаций в половых и соматических клетках
Группы наследственных заболеваний • Гаметические – вызванные вследствие мутаций в половых клетках • Соматические – возникающие вследствие мутаций в соматических клетках • Смешанные – развивающиеся как следствие комбинаций мутаций в половых и соматических клетках
 Частота синдрома среди новорожденных: 1: 700 – 1: 800 Причины") Трисомия 21 (синдром Дауна) Частота синдрома среди новорожденных: 1: 700 – 1: 800 Причины развития: • В 95% случаев – в результате нерасхождения хромосом 21 в гаметах матери или отца, вызванное снижением количества хиазм при первом или втором делении мейоза • В 1 -2% случаев заболеваний наблюдают мозаичный синдром, возникший в результате нарушения сегрегации хромосом в на ранних этапах эмбриогенеза (вероятнее всего, в период дробления) и затрагивающий лишь часть тканей • 2 -3% составляют случаи заболевания, развившиеся в результате Робертсоновских транслокаций в кариотипе одного из родителей – семейный синдром. В этом случае наблюдают прикрепление длинного плеча 21 хромосомы к плечу другой хромосомы (наиболее часто 14) • В очень редких случаях выявляют дублирование части 21 хромосомы.
Трисомия 21 (синдром Дауна) Частота синдрома среди новорожденных: 1: 700 – 1: 800 Причины развития: • В 95% случаев – в результате нерасхождения хромосом 21 в гаметах матери или отца, вызванное снижением количества хиазм при первом или втором делении мейоза • В 1 -2% случаев заболеваний наблюдают мозаичный синдром, возникший в результате нарушения сегрегации хромосом в на ранних этапах эмбриогенеза (вероятнее всего, в период дробления) и затрагивающий лишь часть тканей • 2 -3% составляют случаи заболевания, развившиеся в результате Робертсоновских транслокаций в кариотипе одного из родителей – семейный синдром. В этом случае наблюдают прикрепление длинного плеча 21 хромосомы к плечу другой хромосомы (наиболее часто 14) • В очень редких случаях выявляют дублирование части 21 хромосомы.
 Частота синдрома среди новорожденных: 1: 5000 – 1: 7000 Причины") Трисомия 13 (Синдром Патау) Частота синдрома среди новорожденных: 1: 5000 – 1: 7000 Причины развития: • 75% случаев развития синдрома обусловлено появлением дополнительной хромосомы 13 • В 25% случаев синдром Патау развивается вследствие робертсоновской транслокации с вовлечением 13 хромосомы, в том числе в трех случаях из четырех происходит мутация de novo. В четверти случаев транслокация с вовлечением хромосом 13 -й пары имеет наследственный характер с возвратным риском 14 %. • Другие цитогенетические варианты (мозаицизм, изохромосома, неробертосоновские транслокации) также приводят к развитию синдрома, но встречаются крайне редко. Соотношение полов при синдроме Патау близко к 1: 1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25 — 30 % ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок беременности 38, 5 недель). Большинство детей с синдромом Патау умирают в первые недели и месяцы жизни, лишь 5% живут более 1 года.
Трисомия 13 (Синдром Патау) Частота синдрома среди новорожденных: 1: 5000 – 1: 7000 Причины развития: • 75% случаев развития синдрома обусловлено появлением дополнительной хромосомы 13 • В 25% случаев синдром Патау развивается вследствие робертсоновской транслокации с вовлечением 13 хромосомы, в том числе в трех случаях из четырех происходит мутация de novo. В четверти случаев транслокация с вовлечением хромосом 13 -й пары имеет наследственный характер с возвратным риском 14 %. • Другие цитогенетические варианты (мозаицизм, изохромосома, неробертосоновские транслокации) также приводят к развитию синдрома, но встречаются крайне редко. Соотношение полов при синдроме Патау близко к 1: 1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25 — 30 % ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок беременности 38, 5 недель). Большинство детей с синдромом Патау умирают в первые недели и месяцы жизни, лишь 5% живут более 1 года.
 Частота среди новорожденных: 1: 3500 – 1: 8000 Причины развития:") Трисомия 18 (Синдром Эдвардса) Частота среди новорожденных: 1: 3500 – 1: 8000 Причины развития: • Около 90% случаев составляет нерасхождение хромосом при мейотическом делении • 10% приходится на мозаичные формы синдрома, развившиеся в результате нарушения сегрегации хромосом на ранних этапах эмбриогенеза • Незначительная доля заболеваний возникает в результате транслокаций Для синдрома характерна резкая пренатальная гипоплазия. Дети с синдромом Эдвардса погибают в раннем возрасте. Основной причиной смерти служат остановка дыхания и нарушения работы сердца
Трисомия 18 (Синдром Эдвардса) Частота среди новорожденных: 1: 3500 – 1: 8000 Причины развития: • Около 90% случаев составляет нерасхождение хромосом при мейотическом делении • 10% приходится на мозаичные формы синдрома, развившиеся в результате нарушения сегрегации хромосом на ранних этапах эмбриогенеза • Незначительная доля заболеваний возникает в результате транслокаций Для синдрома характерна резкая пренатальная гипоплазия. Дети с синдромом Эдвардса погибают в раннем возрасте. Основной причиной смерти служат остановка дыхания и нарушения работы сердца
 Синдром триплоидии Частота встречаемости среди новорожденных: Крайне редко – описано меньше 100 случаев Причины развития: • 66% случаев заболевания обусловленны оплодотворением яйцеклетки двумя сперматозоидами (диандрия) • Остальные случаи связаны с нарушениями первого деления мейоза в гаметах отца или матери с образованием сперматозоида (диандрия) или яйцеклетки (дигения) с диплоидным набором Большинство зародышей элиминируется на ранних сроках беременности. Часть детей рождается мертвыми или умирают в первые часы жизни.
Синдром триплоидии Частота встречаемости среди новорожденных: Крайне редко – описано меньше 100 случаев Причины развития: • 66% случаев заболевания обусловленны оплодотворением яйцеклетки двумя сперматозоидами (диандрия) • Остальные случаи связаны с нарушениями первого деления мейоза в гаметах отца или матери с образованием сперматозоида (диандрия) или яйцеклетки (дигения) с диплоидным набором Большинство зародышей элиминируется на ранних сроках беременности. Часть детей рождается мертвыми или умирают в первые часы жизни.
 Частота встречаемости среди новорожденных: 1: 2500 – 1: 5000") Синдром Шерешевского-Тернера (45, Х 0) Частота встречаемости среди новорожденных: 1: 2500 – 1: 5000 Причины развития: • Только 50% случаев синдрома обусловлены истинной моносомией, причем предполагается, что потеря второй половой хромосомы происходит не при мейотическом, а при митотическом делении на ранних стадиях развития • 10% случаев заболевания развиваются в результате таких мутаций как изохромосома, делеция короткого или длинного плеча Х-хромосомы • В остальных случаях наблюдают мозаичную форму синдрома Характерными признаками синдрома Тернера при рождении являются избыток кожи на шее и другие пороки развития, особенно костно-суставной и сердечно-сосудистой систем, «лицо сфинкса» , лимфостаз (застой лимфы, клинически проявляющийся крупными отеками). Интеллект у многих пациентов сохранен, однако частота олигофремнии все же выше.
Синдром Шерешевского-Тернера (45, Х 0) Частота встречаемости среди новорожденных: 1: 2500 – 1: 5000 Причины развития: • Только 50% случаев синдрома обусловлены истинной моносомией, причем предполагается, что потеря второй половой хромосомы происходит не при мейотическом, а при митотическом делении на ранних стадиях развития • 10% случаев заболевания развиваются в результате таких мутаций как изохромосома, делеция короткого или длинного плеча Х-хромосомы • В остальных случаях наблюдают мозаичную форму синдрома Характерными признаками синдрома Тернера при рождении являются избыток кожи на шее и другие пороки развития, особенно костно-суставной и сердечно-сосудистой систем, «лицо сфинкса» , лимфостаз (застой лимфы, клинически проявляющийся крупными отеками). Интеллект у многих пациентов сохранен, однако частота олигофремнии все же выше.
 Синдром кошачьего крика Частота встречаемости среди новорожденных: 1: 45000 Причины развития: Генетически синдром кошачьего крика объясняется частичной моносомией: он развивается при делеции (с утратой от трети до половины, реже полная утрата) короткого плеча пятой хромосомы. Для развития клинической картины синдрома имеет значение не величина утраченного участка, а конкретный незначительный фрагмент хромосомы. Изредка отмечается мозаицизм по делеции или образование кольцевой хромосомы-5. У детей с этой хромосомной аномалией отмечается необычный плач, напоминающий кошачье мяуканье или крик, обусловленный изменением гортани; к году жизни данный признак проходит. Для синдрома характерна задержка умственного и физического развития. Продолжительность жизни варьируется в зависимости от тяжести врожденных пороков развития органов.
Синдром кошачьего крика Частота встречаемости среди новорожденных: 1: 45000 Причины развития: Генетически синдром кошачьего крика объясняется частичной моносомией: он развивается при делеции (с утратой от трети до половины, реже полная утрата) короткого плеча пятой хромосомы. Для развития клинической картины синдрома имеет значение не величина утраченного участка, а конкретный незначительный фрагмент хромосомы. Изредка отмечается мозаицизм по делеции или образование кольцевой хромосомы-5. У детей с этой хромосомной аномалией отмечается необычный плач, напоминающий кошачье мяуканье или крик, обусловленный изменением гортани; к году жизни данный признак проходит. Для синдрома характерна задержка умственного и физического развития. Продолжительность жизни варьируется в зависимости от тяжести врожденных пороков развития органов.
 Синдром Ангельмана –") Микроцитогенетические синдромы Синдром Прадера-Вилли – делеция участка плеча 15 хромосомы (отцовской) Синдром Ангельмана – делеция участка плеча 15 хромосомы (материнской) Синдром Лангера-Гидеона – делеция на 8 хромосоме Опухоль Вильмса – делеция на 11 хромосоме Ретинобластома – делеция на 13 хромосоме Синдром Миллера-Дикера – делеция на 17 хромосоме Синдром Ди Джорджи – делеция на 22 хромосоме Синдром Беквита-Видемана – дупликация на 11 хромосоме
Микроцитогенетические синдромы Синдром Прадера-Вилли – делеция участка плеча 15 хромосомы (отцовской) Синдром Ангельмана – делеция участка плеча 15 хромосомы (материнской) Синдром Лангера-Гидеона – делеция на 8 хромосоме Опухоль Вильмса – делеция на 11 хромосоме Ретинобластома – делеция на 13 хромосоме Синдром Миллера-Дикера – делеция на 17 хромосоме Синдром Ди Джорджи – делеция на 22 хромосоме Синдром Беквита-Видемана – дупликация на 11 хромосоме
 Методы диагностики врожденных заболеваний ØЦитогенетический – хромосомные болезни: уменьшение или увеличение числа хромосом. ØМолекулярно-цитогенетический ØМолекулярно-генетический (например, хорея Гентингтона, миопатия Дюшена, муковисцидоз) ØСиндромологический подход (по фенотипическим признакам)
Методы диагностики врожденных заболеваний ØЦитогенетический – хромосомные болезни: уменьшение или увеличение числа хромосом. ØМолекулярно-цитогенетический ØМолекулярно-генетический (например, хорея Гентингтона, миопатия Дюшена, муковисцидоз) ØСиндромологический подход (по фенотипическим признакам)
 Концентрация хорионического гонадотропина в крови Изменение концентрации хорионического гонадотропина в крови после переноса эмбрионов в программе ЭКО Изменение концентрации хорионического гонадотропина в крови при физиологической беременности Во II триместре беременности, при наличии у плода синдрома Дауна, концентрация ХГЧ в крови беременной женщины повышена. В то же время, повышенная концентрация ХГЧ зачастую наблюдается при многоплодной беременности
Концентрация хорионического гонадотропина в крови Изменение концентрации хорионического гонадотропина в крови после переноса эмбрионов в программе ЭКО Изменение концентрации хорионического гонадотропина в крови при физиологической беременности Во II триместре беременности, при наличии у плода синдрома Дауна, концентрация ХГЧ в крови беременной женщины повышена. В то же время, повышенная концентрация ХГЧ зачастую наблюдается при многоплодной беременности
 Биохимические маркеры трисомий • Свободная β-субъединица ХГЧ. Концентрация свободной β-субъединицы ХГЧ в сыворотке крови беременных очень низка, около 10 недели беременности она максимальна, составляет 1 -3% от всех производных ХГЧ, а к девятому месяцу понижается до 0, 5%. Содержание в крови свободной β-субъединицы значительно увеличивается при синдроме Дауна. Данный маркер является наиболее информативным: уровень β-ХГЧ выше уже в 1 -ом триместре беременности, осложненный трисомией 21. • РАРР-А. Ассоциированный с беременностью плазменный протеин А синтезируется трофобластом. Его концентрация в норме во время беременности постоянно повышается. Уровень этого маркера в конце первого триместра гестации (8 -14 недель) значительно снижен при наличии у плода трисомии 21 или трисомии 18 (синдром Эдвардса). Уникальностью этого показателя является то, что значимость его как маркера синдрома Дауна исчезает после 14 недель беременности. Во втором триместре уровни его в материнской крови при наличии у плода трисомии 21 не отличаются от таковых у беременных со здоровым плодом. Если рассматривать РАРР-А в качестве изолированного маркера риска синдрома Дауна в первом триместре беременности, наиболее значимым было бы его определение в сроки 8 -9 недель. Однако свободная β-субъединица ХГЧ является стабильным маркером риска синдрома Дауна в сроки 10 -18 недель, т. е. позже РАРР-А. Поэтому оптимальным сроком сдачи крови длятеста первого триместра беременности является 10 -12 недель. • Альфа-фетопротеин. В крови матери уровень АФП начинает нарастать с 10 -й недели гестации и достигает максимума к 30 неделям. Уровень АФП в крови матери повышается при открытых дефектах нервной трубке у плода. Наибольшее диагностическое значение имеет определение его содержания в 16 -18 недель. Понижение уровня АФП по сравнению с уровнем нормально развивающейся беременности может свидетельствовать о риске рождения ребенка с трисомией по 21 хромосоме. Однако, поскольку его уровень при патологической беременности составляет лишь 0, 7 от его уровня при физиологической беременности, рекомендуется анализ АФП комбинировать с определением других независимых маркеров – ХГЧ, свободного эстриола. • ХГЧ. Этот гормон синтезируется трофобластом с момента имплантации оплодотворенной яйцеклетки. Во время первого триместра беременности уровень ХГЧ быстро возрастает, удваиваясь каждые 2 -3 дня и достигает максимума на 8 -10 неделе гестации, после чего несколько снижается и остается постоянным в течение второй половины беременности. При синдроме Дауна уровень ХГЧ становится повышенным, при синдроме Эдвардса пониженным
Биохимические маркеры трисомий • Свободная β-субъединица ХГЧ. Концентрация свободной β-субъединицы ХГЧ в сыворотке крови беременных очень низка, около 10 недели беременности она максимальна, составляет 1 -3% от всех производных ХГЧ, а к девятому месяцу понижается до 0, 5%. Содержание в крови свободной β-субъединицы значительно увеличивается при синдроме Дауна. Данный маркер является наиболее информативным: уровень β-ХГЧ выше уже в 1 -ом триместре беременности, осложненный трисомией 21. • РАРР-А. Ассоциированный с беременностью плазменный протеин А синтезируется трофобластом. Его концентрация в норме во время беременности постоянно повышается. Уровень этого маркера в конце первого триместра гестации (8 -14 недель) значительно снижен при наличии у плода трисомии 21 или трисомии 18 (синдром Эдвардса). Уникальностью этого показателя является то, что значимость его как маркера синдрома Дауна исчезает после 14 недель беременности. Во втором триместре уровни его в материнской крови при наличии у плода трисомии 21 не отличаются от таковых у беременных со здоровым плодом. Если рассматривать РАРР-А в качестве изолированного маркера риска синдрома Дауна в первом триместре беременности, наиболее значимым было бы его определение в сроки 8 -9 недель. Однако свободная β-субъединица ХГЧ является стабильным маркером риска синдрома Дауна в сроки 10 -18 недель, т. е. позже РАРР-А. Поэтому оптимальным сроком сдачи крови длятеста первого триместра беременности является 10 -12 недель. • Альфа-фетопротеин. В крови матери уровень АФП начинает нарастать с 10 -й недели гестации и достигает максимума к 30 неделям. Уровень АФП в крови матери повышается при открытых дефектах нервной трубке у плода. Наибольшее диагностическое значение имеет определение его содержания в 16 -18 недель. Понижение уровня АФП по сравнению с уровнем нормально развивающейся беременности может свидетельствовать о риске рождения ребенка с трисомией по 21 хромосоме. Однако, поскольку его уровень при патологической беременности составляет лишь 0, 7 от его уровня при физиологической беременности, рекомендуется анализ АФП комбинировать с определением других независимых маркеров – ХГЧ, свободного эстриола. • ХГЧ. Этот гормон синтезируется трофобластом с момента имплантации оплодотворенной яйцеклетки. Во время первого триместра беременности уровень ХГЧ быстро возрастает, удваиваясь каждые 2 -3 дня и достигает максимума на 8 -10 неделе гестации, после чего несколько снижается и остается постоянным в течение второй половины беременности. При синдроме Дауна уровень ХГЧ становится повышенным, при синдроме Эдвардса пониженным
 Низкий") Интерпретация результатов тройного теста: Нарушение АФП Е 3 ХГ Синдром Дауна (трисомия 21) Низкий Высокий Трисомия 13 Нормальный Нет данных Низкий Трисомия 18 Низкий Открытые дефекты нервной трубки Высокий Нормальный Задержка развития, угроза преждевременных родов, внутриутробная смерть плода Высокий Нет данных Высокий Многоплодная беременность Высокий
Интерпретация результатов тройного теста: Нарушение АФП Е 3 ХГ Синдром Дауна (трисомия 21) Низкий Высокий Трисомия 13 Нормальный Нет данных Низкий Трисомия 18 Низкий Открытые дефекты нервной трубки Высокий Нормальный Задержка развития, угроза преждевременных родов, внутриутробная смерть плода Высокий Нет данных Высокий Многоплодная беременность Высокий
 вид (отношение") Предлежание плода предлежание (головное, тазовое — чисто ягодичное, смешанное и варианты ножного) вид (отношение спинки плода к передней брюшной стенке матери — передний или задний). Головное предлежание плода
Предлежание плода предлежание (головное, тазовое — чисто ягодичное, смешанное и варианты ножного) вид (отношение спинки плода к передней брюшной стенке матери — передний или задний). Головное предлежание плода
 Виды тазового предлежания плода Схематическое изображение положения плода при различных видах тазового предлежания: а — чисто ягодичное (неполное) предлежание (ко входу в малый таз обращены ягодицы плода, ножки согнуты в тазобедренных суставах, разогнуты в коленных суставах и вытянуты вдоль туловища); б — смешанное ягодичное (полное) предлежание (ко входу в малый таз обращены ягодицы и стопы плода, ножки согнуты в тазобедренных и коленных суставах); в — полное ножное предлежание (ко входу в малый таз обращены обе стопы плода); г — неполное ножное предлежание (ко входу в малый таз обращена одна стопа плода).
Виды тазового предлежания плода Схематическое изображение положения плода при различных видах тазового предлежания: а — чисто ягодичное (неполное) предлежание (ко входу в малый таз обращены ягодицы плода, ножки согнуты в тазобедренных суставах, разогнуты в коленных суставах и вытянуты вдоль туловища); б — смешанное ягодичное (полное) предлежание (ко входу в малый таз обращены ягодицы и стопы плода, ножки согнуты в тазобедренных и коленных суставах); в — полное ножное предлежание (ко входу в малый таз обращены обе стопы плода); г — неполное ножное предлежание (ко входу в малый таз обращена одна стопа плода).
 – это состояние, при котором имеет место малое количество") Патология околоплодной среды Маловодие (олигогидрамнион) – это состояние, при котором имеет место малое количество вод, т. е. менее 0, 5 л; или полное их отсутствие – ангидрамнион, встречается в 0, 3 -0, 4% всех родов. При врожденных пороках развития у плодов встречается в 10 раз чаще. Причины: • инфекционно-воспалительные экстрагенитальные и генитальные заболевания матери, • нарушение обменных процессов (ожирение), • органические аномалии мочевыделительной системы плода, • фетоплацентарная недостаточность. Патогенез маловодия – недостаточное развитие эпителия амниона или пониженная его функция. Многоводие (гидрамнион) - патологическое состояние, характеризующееся наличием избыточного количества околоплодных вод в амниотической полости (свыше 1, 5 л). Встречается в 0, 3 -0, 6% всех родов. Может развиваться у беременных, страдающих сахарным диабетом, заболеваниями почек, сердечно-сосудистыми заболеваниями, после инфекционных заболеваний во время беременности, вследствие иммунологической несовместимости крови матери и плода. Развивается при нарушении секреторной и резорбционной функции амниона.
Патология околоплодной среды Маловодие (олигогидрамнион) – это состояние, при котором имеет место малое количество вод, т. е. менее 0, 5 л; или полное их отсутствие – ангидрамнион, встречается в 0, 3 -0, 4% всех родов. При врожденных пороках развития у плодов встречается в 10 раз чаще. Причины: • инфекционно-воспалительные экстрагенитальные и генитальные заболевания матери, • нарушение обменных процессов (ожирение), • органические аномалии мочевыделительной системы плода, • фетоплацентарная недостаточность. Патогенез маловодия – недостаточное развитие эпителия амниона или пониженная его функция. Многоводие (гидрамнион) - патологическое состояние, характеризующееся наличием избыточного количества околоплодных вод в амниотической полости (свыше 1, 5 л). Встречается в 0, 3 -0, 6% всех родов. Может развиваться у беременных, страдающих сахарным диабетом, заболеваниями почек, сердечно-сосудистыми заболеваниями, после инфекционных заболеваний во время беременности, вследствие иммунологической несовместимости крови матери и плода. Развивается при нарушении секреторной и резорбционной функции амниона.
 Плацента – визитная карточка плода
Плацента – визитная карточка плода
 Амниотическая оболочка и в") Плацента – визитная карточка плода I. ПЛОДНАЯ ЧАСТЬ ПЛАЦЕНТЫ а) Амниотическая оболочка и в ней: 1 — однослойный призматический эпителий, 2 — собственная пластинка (плотная волокнистая соединительная ткань). б) Рыхлая ("слизистая") соединительная ткань (3) между амнионом и хорионом. в) Ветвистый хорион: 4 А — хориальная пластинка и отходящие от нее 4 Б — стволовые ворсины. Одна такая ворсина со всеми ее разветвлениями называется котиледоном. Компоненты хориона (в пластинке и в ворсинах): 5 — соединительная ткань; 6 — ветви пупочных сосудов; эпителий (на поверхности ворсин): 7 — цитотрофобласт (внутренний слой клеток); 8 — симпластотрофобласт; отсутствует в местах контакта якорных ворсин с базальной пластинкой эндометрия; фибриноид Лангханса — неклеточная фибриноподобная масса на поверхности эпителия (появляется со второй половины беременности). II. МАТЕРИНСКАЯ ЧАСТЬ ПЛАЦЕНТЫ (decidua basalis) 9 — лакуны и в них: 10 — материнская кровь; 11 —соединительнотканные септы (перегородки) между лакунами; 12 — сосуды матери в септах, открывающиеся в лакуны; 13 — базальная пластинка; включает соединительную ткань и 14 — скопления децидуальных клеток. Клетки — крупные, светлые, обладают макрофагальной активностью. Последняя резко возрастает перед родами и способствует отторжению плаценты.
Плацента – визитная карточка плода I. ПЛОДНАЯ ЧАСТЬ ПЛАЦЕНТЫ а) Амниотическая оболочка и в ней: 1 — однослойный призматический эпителий, 2 — собственная пластинка (плотная волокнистая соединительная ткань). б) Рыхлая ("слизистая") соединительная ткань (3) между амнионом и хорионом. в) Ветвистый хорион: 4 А — хориальная пластинка и отходящие от нее 4 Б — стволовые ворсины. Одна такая ворсина со всеми ее разветвлениями называется котиледоном. Компоненты хориона (в пластинке и в ворсинах): 5 — соединительная ткань; 6 — ветви пупочных сосудов; эпителий (на поверхности ворсин): 7 — цитотрофобласт (внутренний слой клеток); 8 — симпластотрофобласт; отсутствует в местах контакта якорных ворсин с базальной пластинкой эндометрия; фибриноид Лангханса — неклеточная фибриноподобная масса на поверхности эпителия (появляется со второй половины беременности). II. МАТЕРИНСКАЯ ЧАСТЬ ПЛАЦЕНТЫ (decidua basalis) 9 — лакуны и в них: 10 — материнская кровь; 11 —соединительнотканные септы (перегородки) между лакунами; 12 — сосуды матери в септах, открывающиеся в лакуны; 13 — базальная пластинка; включает соединительную ткань и 14 — скопления децидуальных клеток. Клетки — крупные, светлые, обладают макрофагальной активностью. Последняя резко возрастает перед родами и способствует отторжению плаценты.
 Трофобластическая болезнь -группа доброкачественных и злокачественных новообразований происходящих из элементов плаценты. Встречается относительно редко: на 1000 родов отмечается 1 наблюдение пузырного заноса, на 100. 000 родов или абортов приходится 2 случая хориокарциномы. Cогласно Международной классификацией онкологических болезней (1995) к трофобластической болезни относятся: • Пузырный занос (полный или частичный). • Инвазивный пузырный занос. • Хориокарцинома или хорионэпителиома. • Хориокарцинома в сочетании с тератомой или эмбриональным раком. • Злокачественная тератома трофобластическая. • Трофобластическая опухоль плацентарной площадки. Причины трофобластической болезни: вирусная теория, иммунологическая теория, ферментативная теория, дефицит белка. Факторы риска: возраст пациенток старше 40 лет (риск заболевания в 5 раз выше, чем для женщин в возрасте от 21 до 35 лет); наличие в анамнезе ранних самопроизвольных абортов; вероятность заболевания выше у повторнобеременных; заболевание чаще встречается в странах Востока по сравнению с западными странами
Трофобластическая болезнь -группа доброкачественных и злокачественных новообразований происходящих из элементов плаценты. Встречается относительно редко: на 1000 родов отмечается 1 наблюдение пузырного заноса, на 100. 000 родов или абортов приходится 2 случая хориокарциномы. Cогласно Международной классификацией онкологических болезней (1995) к трофобластической болезни относятся: • Пузырный занос (полный или частичный). • Инвазивный пузырный занос. • Хориокарцинома или хорионэпителиома. • Хориокарцинома в сочетании с тератомой или эмбриональным раком. • Злокачественная тератома трофобластическая. • Трофобластическая опухоль плацентарной площадки. Причины трофобластической болезни: вирусная теория, иммунологическая теория, ферментативная теория, дефицит белка. Факторы риска: возраст пациенток старше 40 лет (риск заболевания в 5 раз выше, чем для женщин в возрасте от 21 до 35 лет); наличие в анамнезе ранних самопроизвольных абортов; вероятность заболевания выше у повторнобеременных; заболевание чаще встречается в странах Востока по сравнению с западными странами
 Пузырный занос - патологически измененный ворсинчатый хорион в виде множества пузырьков различной величины, наполненных прозрачной жидкостью, которые полностью (полный занос) или частично (частичный занос) замещают ткань плаценты. Элементы пузырного заноса могут свободно находиться в полости матки и быть связанными с ее стенкой. Хорионкарцинома (синоним - хорионэпителиома) - злокачественная форма трофобластической болезни, возникающая из хориального эпителия после пузырного заноса (до 40% случаев), после нормального аборта (25%) или после родов (22, 5%). Диагностика трофобластической болезни: • Сбор анамнеза и гинекологический осмотр с учетом жалоб пациентки • УЗИ • Цветное доплеровское картирование • Рентгенологическое исследование • Гистероскопия • Тазовая ангиография
Пузырный занос - патологически измененный ворсинчатый хорион в виде множества пузырьков различной величины, наполненных прозрачной жидкостью, которые полностью (полный занос) или частично (частичный занос) замещают ткань плаценты. Элементы пузырного заноса могут свободно находиться в полости матки и быть связанными с ее стенкой. Хорионкарцинома (синоним - хорионэпителиома) - злокачественная форма трофобластической болезни, возникающая из хориального эпителия после пузырного заноса (до 40% случаев), после нормального аборта (25%) или после родов (22, 5%). Диагностика трофобластической болезни: • Сбор анамнеза и гинекологический осмотр с учетом жалоб пациентки • УЗИ • Цветное доплеровское картирование • Рентгенологическое исследование • Гистероскопия • Тазовая ангиография
 Вредные воздействия на ранних этапах эмбрионального развития приводят к грубым нарушениям морфогенеза - эмбриопатиям Конюхов, 1980
Вредные воздействия на ранних этапах эмбрионального развития приводят к грубым нарушениям морфогенеза - эмбриопатиям Конюхов, 1980
 SAMP 1") Мыши линий SAM (мыши, склонные к ускоренному старению – Senescense Accelerated Mouse) SAMP 1 Senescence Accelerated Mouse Prone SAMR 1 Senescence Accelerated Mouse Resistant животные в возрасте 11 месяцев
Мыши линий SAM (мыши, склонные к ускоренному старению – Senescense Accelerated Mouse) SAMP 1 Senescence Accelerated Mouse Prone SAMR 1 Senescence Accelerated Mouse Resistant животные в возрасте 11 месяцев
 Эмбрионы и неоплодотворенные ооциты SAMP 1 1. 5 сутки развития норма + кит LIVE/DEAD фрагментация дегенерирующие неоплодотворенные ооциты + кит LIVE/DEAD
Эмбрионы и неоплодотворенные ооциты SAMP 1 1. 5 сутки развития норма + кит LIVE/DEAD фрагментация дегенерирующие неоплодотворенные ооциты + кит LIVE/DEAD
 Эмбрионы SAMP 1 2. 5 сутки развития норма мало клеток фрагментация неполная два центра компактизации нарушения компактизации
Эмбрионы SAMP 1 2. 5 сутки развития норма мало клеток фрагментация неполная два центра компактизации нарушения компактизации
 Эмбрионы SAMP 1 норма нет внутренней клеточной массы 3. 5 сутки развития нарушения кавитации две полости мало клеток в трофэктодерме два центра компактизации нарушения компактизации и кавитация из малого числа клеток
Эмбрионы SAMP 1 норма нет внутренней клеточной массы 3. 5 сутки развития нарушения кавитации две полости мало клеток в трофэктодерме два центра компактизации нарушения компактизации и кавитация из малого числа клеток
 Распределение эмбрионов SAMP 1 по морфологическим группам 1. 5 суток развития 2. 5 суток развития 18 самок, 140 эмбрионов 51 самка, 370 эмбрионов остановка развития в период дробления дегенерация норма фрагментация 3. 5 суток развития фрагментация 27 самок, 244 эмбриона нарушение кавитации нарушение компактизации норма * p < 0. 001 vs. 1. 5 и 2. 5 фрагментация отставание в развитии дегенерация остановка развития в период дробления норма
Распределение эмбрионов SAMP 1 по морфологическим группам 1. 5 суток развития 2. 5 суток развития 18 самок, 140 эмбрионов 51 самка, 370 эмбрионов остановка развития в период дробления дегенерация норма фрагментация 3. 5 суток развития фрагментация 27 самок, 244 эмбриона нарушение кавитации нарушение компактизации норма * p < 0. 001 vs. 1. 5 и 2. 5 фрагментация отставание в развитии дегенерация остановка развития в период дробления норма
 Распределение эмбрионов по морфологическим группам на 2. 5 сутки линии SAMP 1 51 самка, 370 эмбрионов остановка развития в период дробления SAMR 1 25 самок, 131 эмбрион остановка развития в период дробления * p<0. 048 дегенерация *** p<0. 012 норма x p<0. 001 норма дегенерация фрагментация ** p<0. 039 фрагментация линии с нормальной плодовитостью ICR 19 самок, 144 эмбриона остановка развития в период дробления дегенерация фрагментация остановка развития в период дробления 51 самка, 358 эмбрионов дегенерация норма x, *, BALB/DBA **, *** – сравнение линий SAM и линий с нормальной плодовитостью норма
Распределение эмбрионов по морфологическим группам на 2. 5 сутки линии SAMP 1 51 самка, 370 эмбрионов остановка развития в период дробления SAMR 1 25 самок, 131 эмбрион остановка развития в период дробления * p<0. 048 дегенерация *** p<0. 012 норма x p<0. 001 норма дегенерация фрагментация ** p<0. 039 фрагментация линии с нормальной плодовитостью ICR 19 самок, 144 эмбриона остановка развития в период дробления дегенерация фрагментация остановка развития в период дробления 51 самка, 358 эмбрионов дегенерация норма x, *, BALB/DBA **, *** – сравнение линий SAM и линий с нормальной плодовитостью норма
 ↓ ICR нарушения компактизации и") Эмбрионы после культивирования с блеомицином (1 мкг/мл, 48 часов) ↓ ICR нарушения компактизации и кавитация из малого числа клеток + кит LIVE/DEAD мало клеток в эмбрионе ↑ SAMP 1 + кит LIVE/DEAD две внутренние клеточные массы нарушения компактизации и кавитации две полости
Эмбрионы после культивирования с блеомицином (1 мкг/мл, 48 часов) ↓ ICR нарушения компактизации и кавитация из малого числа клеток + кит LIVE/DEAD мало клеток в эмбрионе ↑ SAMP 1 + кит LIVE/DEAD две внутренние клеточные массы нарушения компактизации и кавитации две полости
 Рафаэль Санти «Мадонна в кресле»
Рафаэль Санти «Мадонна в кресле»