

Афонников6_2011.ppt
- Количество слайдов: 62
 Филогенетические деревья Корень Время Порядок ветвления – точки на шкале времени • Необходимы для восстановления эволюционной истории • Описывают эволюционные отношения для набора из нескольких последовательностей
Филогенетические деревья Корень Время Порядок ветвления – точки на шкале времени • Необходимы для восстановления эволюционной истории • Описывают эволюционные отношения для набора из нескольких последовательностей
, соответствуют таксономическим") Филогенетические деревья Конечные узлы Внутренние узлы Корень A-E – конечные узлы (листья), соответствуют таксономическим единицам (OTU); F-I внутренние узлы (предковые) Таксономические единицы: виды, популяции, особи, гены, белки. Потомки эволюционируют независимо. Топология – порядок ветвления узлов дерева.
Филогенетические деревья Конечные узлы Внутренние узлы Корень A-E – конечные узлы (листья), соответствуют таксономическим единицам (OTU); F-I внутренние узлы (предковые) Таксономические единицы: виды, популяции, особи, гены, белки. Потомки эволюционируют независимо. Топология – порядок ветвления узлов дерева.
 Не все деревья имеют корень i, j, - внутренние узлы, последовательности для которых неизвестны • Не все методы построения деревьев могут давать положение корня
Не все деревья имеют корень i, j, - внутренние узлы, последовательности для которых неизвестны • Не все методы построения деревьев могут давать положение корня
 Основанные на наблюдаемых") Группы методов построения деревьев Основанные на эволюционных расстояниях (UPGMA, ближайшего соседа) Основанные на наблюдаемых признаках нуклеотидах, аминокислотах (метод максимальной экономии, максимального правдоподобия).
Группы методов построения деревьев Основанные на эволюционных расстояниях (UPGMA, ближайшего соседа) Основанные на наблюдаемых признаках нуклеотидах, аминокислотах (метод максимальной экономии, максимального правдоподобия).
 Методы построения деревьев: UPGMA • Расстояние между кластером X и кластером Y равно среднему от парных расстояний между последовательностя ми этих кластеров • Предполагает равномерность замен (молек. часы) во всех таксонах • Расстояние=2*длину ветви • Дает всегда дерево с корнем
Методы построения деревьев: UPGMA • Расстояние между кластером X и кластером Y равно среднему от парных расстояний между последовательностя ми этих кластеров • Предполагает равномерность замен (молек. часы) во всех таксонах • Расстояние=2*длину ветви • Дает всегда дерево с корнем
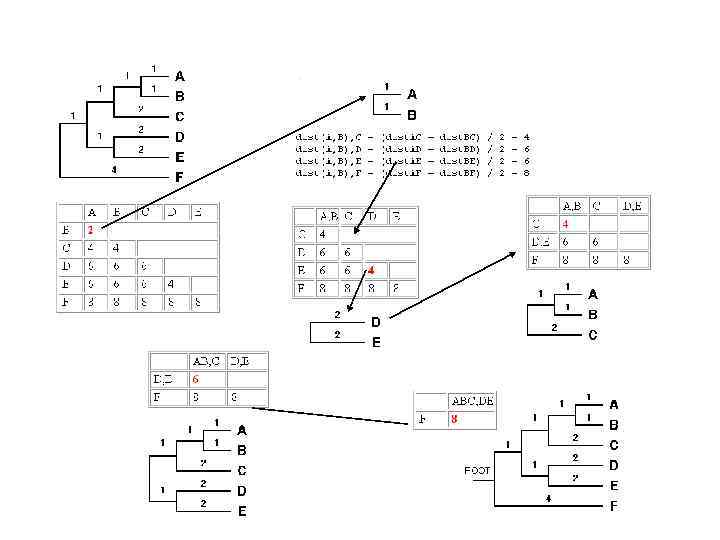
 UPGMA ошибается если скорости замен различны
UPGMA ошибается если скорости замен различны
 Метод ближайшего соседа В этом подходе расстояния нормируются на среднюю удаленность ri OTU от других OTU При построении дерева два ближайших узла i, j заменяются новым узлом n; расстояния пересчитываются по следующим правилам: Для каждого к На следующем шаге выбирается пара i, j, для которых Dij минимально ( Dij=dij-ri-rj )
Метод ближайшего соседа В этом подходе расстояния нормируются на среднюю удаленность ri OTU от других OTU При построении дерева два ближайших узла i, j заменяются новым узлом n; расстояния пересчитываются по следующим правилам: Для каждого к На следующем шаге выбирается пара i, j, для которых Dij минимально ( Dij=dij-ri-rj )
 Для пары A, B Средняя удаленность Первый шаг – стартуем с звездного дерева Второй шаг - A, B образуют новую единицу, U U
Для пары A, B Средняя удаленность Первый шаг – стартуем с звездного дерева Второй шаг - A, B образуют новую единицу, U U
, • Clustal.") Программы реализующие данных подход • Neighbor в пакете Phylip (Felsentein, Univ. Washington), • Clustal. W (D. Higgins), • Distnj в пакете Protml (Adachi and Hasegawa, Univ. Tokyo) Учитывает неравномерность скоростей эволюции на ветвях Быстрый, может использоваться для больших семейств Иногда могут встречаться отрицательные расстояния.
Программы реализующие данных подход • Neighbor в пакете Phylip (Felsentein, Univ. Washington), • Clustal. W (D. Higgins), • Distnj в пакете Protml (Adachi and Hasegawa, Univ. Tokyo) Учитывает неравномерность скоростей эволюции на ветвях Быстрый, может использоваться для больших семейств Иногда могут встречаться отрицательные расстояния.
 Дерево можно перестраивать и оценивать Перестановка соседних узлов Число различных топологий дерева Без корня С корнем Сокращение и перестройка поддеревьев • Каждому дереву можно присвоить числовую характеристику и сравнивать их
Дерево можно перестраивать и оценивать Перестановка соседних узлов Число различных топологий дерева Без корня С корнем Сокращение и перестройка поддеревьев • Каждому дереву можно присвоить числовую характеристику и сравнивать их
 Метод парсимонии Пример построения дерева для набора из четырех последовательностей, с двузнаковым алфавитом (0, 1) 1) В первом дереве изменения происходят только один раз (+) 2) Во втором дереве 1 появляется (+) и теряется (*) 3) В третьем дереве 1 появляется независимо два раза (+) Дерево (1) содержит минимальное число эволюционных событий – его и выбираем.
Метод парсимонии Пример построения дерева для набора из четырех последовательностей, с двузнаковым алфавитом (0, 1) 1) В первом дереве изменения происходят только один раз (+) 2) Во втором дереве 1 появляется (+) и теряется (*) 3) В третьем дереве 1 появляется независимо два раза (+) Дерево (1) содержит минимальное число эволюционных событий – его и выбираем.
 • Paup (David Swofford) Приемлем") Программы реализующие данных подход • Protpars (Felsentein, пакет Phylip) • Paup (David Swofford) Приемлем для последовательностей с высокой гомологией. Нельзя использовать для сильно дивергировавших последовательностей!
Программы реализующие данных подход • Protpars (Felsentein, пакет Phylip) • Paup (David Swofford) Приемлем для последовательностей с высокой гомологией. Нельзя использовать для сильно дивергировавших последовательностей!
 Имеются n наблюдений случайной величины x – вектор наблюдений x=(x 1,") Функция правдоподобия (ФП) Имеются n наблюдений случайной величины x – вектор наблюдений x=(x 1, x 2, …xn); Вероятность наблюдать значение x зависит от некоторого параметра : p(x| ). Тогда вероятность наблюдать n значений x=(x 1, x 2, …xn) равна L(x| )=p(x 1| ) p(x 2| ) …. p(xn| ) L(X| ) называют функцией правдоподобия. Ее удобно использовать при оценке параметров распределений p(x| ). Идея: выбрать такой параметр, который максимизирует вероятность наблюдать набор значений x=(x 1, x 2, …xn).
Функция правдоподобия (ФП) Имеются n наблюдений случайной величины x – вектор наблюдений x=(x 1, x 2, …xn); Вероятность наблюдать значение x зависит от некоторого параметра : p(x| ). Тогда вероятность наблюдать n значений x=(x 1, x 2, …xn) равна L(x| )=p(x 1| ) p(x 2| ) …. p(xn| ) L(X| ) называют функцией правдоподобия. Ее удобно использовать при оценке параметров распределений p(x| ). Идея: выбрать такой параметр, который максимизирует вероятность наблюдать набор значений x=(x 1, x 2, …xn).
 – p, вероятность решки (Р)") Пример : бросание монетки Бросаем монету, вероятность орла (O) – p, вероятность решки (Р) – 1 -p. В данном случае параметр, от которого зависит вероятность наблюдать событие O – p. Наблюдаем 11 бросаний монет: ООРОРРО Функция правдоподобия: L=pp(1 − p)(1 − p)p(1 − p)p L=p 5(1 − p)6 L(p)
Пример : бросание монетки Бросаем монету, вероятность орла (O) – p, вероятность решки (Р) – 1 -p. В данном случае параметр, от которого зависит вероятность наблюдать событие O – p. Наблюдаем 11 бросаний монет: ООРОРРО Функция правдоподобия: L=pp(1 − p)(1 − p)p(1 − p)p L=p 5(1 − p)6 L(p)
") Оценка параметра p Обычно используют логарифм Ф. П. (логарифмирование не меняет положение максимума)
Оценка параметра p Обычно используют логарифм Ф. П. (логарифмирование не меняет положение максимума)
 Если много параметров В случае нескольких параметров определяется поверхность правдоподобия ОМП контур
Если много параметров В случае нескольких параметров определяется поверхность правдоподобия ОМП контур
 Функция правдоподобия и проверка гипотез Данные Гипотеза 1 Гипотеза 2 Условная вероятность Отношение постериорных вероятностей Отношение правдоподобия Отношение априорных вероятностей Выбирается гипотеза, при которой вероятность наблюдать набор данных выше (отношение априорных вероятностей полагают 1).
Функция правдоподобия и проверка гипотез Данные Гипотеза 1 Гипотеза 2 Условная вероятность Отношение постериорных вероятностей Отношение правдоподобия Отношение априорных вероятностей Выбирается гипотеза, при которой вероятность наблюдать набор данных выше (отношение априорных вероятностей полагают 1).
 Пример: тест на модель частот нуклеотидов Пусть имеется последовательность нуклеотидов, в которых частоты их встречаемости A C G T Гипотеза Н 1: частоты встречаемости A C G T равны их оценкам в последовательности Гипотеза Н 2: частоты встречаемости A = C= G = T =1/4 (модель Джукса-Кантора)
Пример: тест на модель частот нуклеотидов Пусть имеется последовательность нуклеотидов, в которых частоты их встречаемости A C G T Гипотеза Н 1: частоты встречаемости A C G T равны их оценкам в последовательности Гипотеза Н 2: частоты встречаемости A = C= G = T =1/4 (модель Джукса-Кантора)
 Сравнение значений ФП Гипотеза 1 Гипотеза 2 Принять гипотезу 1 выгоднее, первая модель более вероятна.
Сравнение значений ФП Гипотеза 1 Гипотеза 2 Принять гипотезу 1 выгоднее, первая модель более вероятна.
 Пример сравнения двух последовательностей Сравниваются две последовательности, эволюционировавшие в течении времени t со скоростью замен t Простейший случай – 2 нуклеотида, модель Джукса-Кантора
Пример сравнения двух последовательностей Сравниваются две последовательности, эволюционировавшие в течении времени t со скоростью замен t Простейший случай – 2 нуклеотида, модель Джукса-Кантора
, при условии, что эволюция происходила") Методы максимального правдоподобия • Вычисляется вероятность наблюдения данных (D), при условии, что эволюция происходила по данной топологии (T) – функция правдоподобия L(D|T) • Вычисляется рекурсивно от листьев к вершине • Выбирается дерево, которое дает max(L) • Решение зависит от выбора модели замен
Методы максимального правдоподобия • Вычисляется вероятность наблюдения данных (D), при условии, что эволюция происходила по данной топологии (T) – функция правдоподобия L(D|T) • Вычисляется рекурсивно от листьев к вершине • Выбирается дерево, которое дает max(L) • Решение зависит от выбора модели замен
; Задана топология дерева (порядок") Как оценить правдоподобие дерева? Задана модель эволюции (матрица скоростей замен); Задана топология дерева (порядок ветвления) и длины ветвей (ti=скорость замен * время)
Как оценить правдоподобие дерева? Задана модель эволюции (матрица скоростей замен); Задана топология дерева (порядок ветвления) и длины ветвей (ti=скорость замен * время)
 Если бы внутренние узлы были известны
Если бы внутренние узлы были известны
") Но они неизвестны Необходимо просуммировать по всем нуклеотидам во внутренних узлах дерева (усреднить)
Но они неизвестны Необходимо просуммировать по всем нуклеотидам во внутренних узлах дерева (усреднить)
 Таксон j A") Подсчет методом сокращения Felsenstein, 1981 (пример для одной позиции выравненных последовательностей) Таксон j A T G C Lj(A)Lj(T) Lj(G) Lj(C) Ветвь ij, Эвол. Расст. vj A T G C Предковый узел i Таксон k A T G C Lk(A) Lk(T)Lk(G) Lk(C) Ветвь ik, Эвол. Расст. vk Prob(sj|s, vj) – Вероятность наблюдать нуклеотид типа sj в дочернем узле j при условии, что в родительском узле i находится символ s и время эволюции составило vj.
Подсчет методом сокращения Felsenstein, 1981 (пример для одной позиции выравненных последовательностей) Таксон j A T G C Lj(A)Lj(T) Lj(G) Lj(C) Ветвь ij, Эвол. Расст. vj A T G C Предковый узел i Таксон k A T G C Lk(A) Lk(T)Lk(G) Lk(C) Ветвь ik, Эвол. Расст. vk Prob(sj|s, vj) – Вероятность наблюдать нуклеотид типа sj в дочернем узле j при условии, что в родительском узле i находится символ s и время эволюции составило vj.
 общей предковой последовательности L 0(i) Итоговое значение") Итоговое значение усредняется по частотам нуклеотидов (аминокислот) общей предковой последовательности L 0(i) Итоговое значение Ф. П. : перемножаются для всех позиций (независимость мутаций в позициях)
Итоговое значение усредняется по частотам нуклеотидов (аминокислот) общей предковой последовательности L 0(i) Итоговое значение Ф. П. : перемножаются для всех позиций (независимость мутаций в позициях)
 • Fast. DNAML (ДНК)") Программы реализующие данных подход • DNAML (пакет Phylip, ДНК ) • Fast. DNAML (ДНК) • Prot. ML (ДНК и белки, Adachi and Hasegawa) • Puzzle (Днк и белки, Strimmer and von Haeseler) • Phyml (ДНК, белки ; Guindon, Gasquel) • Ra. XML (ДНК, белки (если очень много последовательностей – более 1000, быстрый эффективный), Stamatakis)
Программы реализующие данных подход • DNAML (пакет Phylip, ДНК ) • Fast. DNAML (ДНК) • Prot. ML (ДНК и белки, Adachi and Hasegawa) • Puzzle (Днк и белки, Strimmer and von Haeseler) • Phyml (ДНК, белки ; Guindon, Gasquel) • Ra. XML (ДНК, белки (если очень много последовательностей – более 1000, быстрый эффективный), Stamatakis)
 Методы максимального правдоподобия • С помощью методов МП можно оценивать и другие параметры: так как и матрица замен, и скорости замен могут быть такими параметрами. L=L(T, M, t…. . ). • Можно усложнять модель, добавляя новые параметры. • Метод имеет статистическое обоснование • Но требует большого количества вычислений Матрица замен, оцененная методом МП – WAG [Whealan and Goldman] (лучше Dayhoff, JTT).
Методы максимального правдоподобия • С помощью методов МП можно оценивать и другие параметры: так как и матрица замен, и скорости замен могут быть такими параметрами. L=L(T, M, t…. . ). • Можно усложнять модель, добавляя новые параметры. • Метод имеет статистическое обоснование • Но требует большого количества вычислений Матрица замен, оцененная методом МП – WAG [Whealan and Goldman] (лучше Dayhoff, JTT).
 Стабилизирующий отбор (Purifying selection) ; (b)") Дарвиновская идея естественного отбора Три режима отбора: (a) Стабилизирующий отбор (Purifying selection) ; (b) Движущий (Дарвиновский) отбор (Positive selection); (c) Нейтральная эволюция. Приспособленность Доля особей Значение характеристики (a) Приспособленность Доля особей Значение характеристики (б) Приспособленность Доля особей Значение характеристики (в)
Дарвиновская идея естественного отбора Три режима отбора: (a) Стабилизирующий отбор (Purifying selection) ; (b) Движущий (Дарвиновский) отбор (Positive selection); (c) Нейтральная эволюция. Приспособленность Доля особей Значение характеристики (a) Приспособленность Доля особей Значение характеристики (б) Приспособленность Доля особей Значение характеристики (в)
 Нейтральная теория Кимуры Нейтральные мутации могут фиксироваться в популяции стохастическим образом. Пример схематически показан на диаграмме: Адаптивные мутации Число копий гена Неблагоприятные мутации Время фиксации нейтральных мутаций tfix=4 N поколений Скорость фиксации нейтральных замен (число фиксаций за единицу времени) K=u, u- частота возникновения мутации Для адаптивных замен K=4 Nsu
Нейтральная теория Кимуры Нейтральные мутации могут фиксироваться в популяции стохастическим образом. Пример схематически показан на диаграмме: Адаптивные мутации Число копий гена Неблагоприятные мутации Время фиксации нейтральных мутаций tfix=4 N поколений Скорость фиксации нейтральных замен (число фиксаций за единицу времени) K=u, u- частота возникновения мутации Для адаптивных замен K=4 Nsu
 Частота нейтральных мутаций в белках Распределение мутаций до отбора Распределение мутаций после отбора • Адаптивные мутации очень редки • Деструктивные мутации быстро элиминируются • Подавляющее большинство фиксировавшихся мутаций селективно нейтральны • Скорость фиксации замен в генах является постоянной (гипотеза молекулярных часов, Zukercandle & Poling • Функционально важные районы функционируют медленнее, менее важные - быстрее Рисунок из http: //nitro. biosci. arizona. edu/courses/EEB 320/Lecture 46. html
Частота нейтральных мутаций в белках Распределение мутаций до отбора Распределение мутаций после отбора • Адаптивные мутации очень редки • Деструктивные мутации быстро элиминируются • Подавляющее большинство фиксировавшихся мутаций селективно нейтральны • Скорость фиксации замен в генах является постоянной (гипотеза молекулярных часов, Zukercandle & Poling • Функционально важные районы функционируют медленнее, менее важные - быстрее Рисунок из http: //nitro. biosci. arizona. edu/courses/EEB 320/Lecture 46. html
 Являются ли замены адаптивными? Ответ на этот вопрос позволит совершенствовать модели эволюции и выявлять гены которые являются функционально важными. Для кодирующих частей генов существует критерий, который основан на особенностях генетического кода.
Являются ли замены адаптивными? Ответ на этот вопрос позволит совершенствовать модели эволюции и выявлять гены которые являются функционально важными. Для кодирующих частей генов существует критерий, который основан на особенностях генетического кода.
 Генетический код Рисунок Darryl Leja http: //www. accessexcellence. org/AB/GG/codon. html Рисунок из Молекулярная биология © 2000 Дымшиц Г. М. http: //www. nsu. ru/education/biology/molbiol/
Генетический код Рисунок Darryl Leja http: //www. accessexcellence. org/AB/GG/codon. html Рисунок из Молекулярная биология © 2000 Дымшиц Г. М. http: //www. nsu. ru/education/biology/molbiol/
 Вырожденность замен в кодоне Leu UUA UUG CUU CUC CUA CUG кодоны CUA CUG CUC CGU CCU CAU UUU AUU GUU 1/3 - синонимические 2/3 - несинонимические 9 возможных замен • При условии равновероятности замен и одинаковой частоты встречаемости нуклеотидов - 5% замен по 1 позиции и 72% замен по 3 позиции - синонимические.
Вырожденность замен в кодоне Leu UUA UUG CUU CUC CUA CUG кодоны CUA CUG CUC CGU CCU CAU UUU AUU GUU 1/3 - синонимические 2/3 - несинонимические 9 возможных замен • При условии равновероятности замен и одинаковой частоты встречаемости нуклеотидов - 5% замен по 1 позиции и 72% замен по 3 позиции - синонимические.
 • Молчащие замены: Sequence 1: UUU CAU CGU Sequence 2: UUU CAC CGU Аминокислоты: Phe His l. Замены Arg аминокислот: Sequence 1: UUU CAU CGU Sequence 2: UUU CAG CGU Аминокислоты: Phe His Arg Gln
• Молчащие замены: Sequence 1: UUU CAU CGU Sequence 2: UUU CAC CGU Аминокислоты: Phe His l. Замены Arg аминокислот: Sequence 1: UUU CAU CGU Sequence 2: UUU CAG CGU Аминокислоты: Phe His Arg Gln
 Как определить положительный отбор? • dn – скорость несинонимических замен Число несинонимических замен число кодонов с заменами а. к. • ds - скорость синонимических замен Число кодонов без замен а. к. =dn/ds > 1 - положительный отбор (Другое обозначение скоростей – Ka, Ks)
Как определить положительный отбор? • dn – скорость несинонимических замен Число несинонимических замен число кодонов с заменами а. к. • ds - скорость синонимических замен Число кодонов без замен а. к. =dn/ds > 1 - положительный отбор (Другое обозначение скоростей – Ka, Ks)
 Как определить режим отбора? • = 1: • < 1: • > 1: нейтральная эволюция отрицательный(стабилизирующий) отбор положительный (движущий) отбор Этот тест относится только к кодирующим частям генов и может быть неверным, если отбор идет по другим признакам (например, поддержание вторичной структуры РНК)
Как определить режим отбора? • = 1: • < 1: • > 1: нейтральная эволюция отрицательный(стабилизирующий) отбор положительный (движущий) отбор Этот тест относится только к кодирующим частям генов и может быть неверным, если отбор идет по другим признакам (например, поддержание вторичной структуры РНК)
 Методы оценки синонимических и несинонимических замен Существует два основных подхода к оценке dn и ds 1) Метод эволюционного расстояния (Nei, Gojobori (1986) и похожие алгоритмы) Программы: MEGA, CRANN 2) Метод максимального правдоподобия (Yang и сотр), Программа PAML (подпрограмма codeml)
Методы оценки синонимических и несинонимических замен Существует два основных подхода к оценке dn и ds 1) Метод эволюционного расстояния (Nei, Gojobori (1986) и похожие алгоритмы) Программы: MEGA, CRANN 2) Метод максимального правдоподобия (Yang и сотр), Программа PAML (подпрограмма codeml)
 Методы оценки синонимических и несинонимических замен Постановка задачи. Существуют две нуклеотидные последовательности, выровненные так, что они совмещены по кодонам. Требуется оценить скорости синонимических и несинонимических замен и их отношение. S- число синонимических позиций в последовательностях (число всех одиночных нукл. замен, которые синонимические) N –число несинонимических позиций в последовательности (число всех одиночных нукл. замен, которые несинонимические) Sd- число синонимических позиций в которых произошли замены Nd –число несинонимических позиций в которых произошли замены S (пространство синонимических замен) Sd (свершившиеся замены) Sd и Nd напрямую сравнивать нельзя, так как S и N неравны Поэтому сравнивают Sd/S и Nd/N N (пространство несинонимических замен) Nd (свершившиеся замены)
Методы оценки синонимических и несинонимических замен Постановка задачи. Существуют две нуклеотидные последовательности, выровненные так, что они совмещены по кодонам. Требуется оценить скорости синонимических и несинонимических замен и их отношение. S- число синонимических позиций в последовательностях (число всех одиночных нукл. замен, которые синонимические) N –число несинонимических позиций в последовательности (число всех одиночных нукл. замен, которые несинонимические) Sd- число синонимических позиций в которых произошли замены Nd –число несинонимических позиций в которых произошли замены S (пространство синонимических замен) Sd (свершившиеся замены) Sd и Nd напрямую сравнивать нельзя, так как S и N неравны Поэтому сравнивают Sd/S и Nd/N N (пространство несинонимических замен) Nd (свершившиеся замены)
 si – доля синонимических мутаций кодона i ni – доля несинонимических мутаций кодона i Например для кодона CUU - si =1/3; ni = 2/3 S+N=3 r, где r – число кодонов. sdi – доля синонимических замен кодона i ndi – доля несинонимических замен кодона i
si – доля синонимических мутаций кодона i ni – доля несинонимических мутаций кодона i Например для кодона CUU - si =1/3; ni = 2/3 S+N=3 r, где r – число кодонов. sdi – доля синонимических замен кодона i ndi – доля несинонимических замен кодона i
 -> GUA (Val) sd=1; nd=0. Случай двух") Подсчет числа замен. Случай одной замены: GUU(Val) -> GUA (Val) sd=1; nd=0. Случай двух замен: Рассматриваются несколько возможных путей, которые считаются равновероятными. UUU(Phe) -> GUA(Val) : (1): UUU(Phe) -> GUU(Val) -> GUA(Val) sd=1; nd=1. (2): UUU(Phe) -> UUA(Leu) -> GUA(Val) sd=0; nd=2. (1)+(2): sd=0. 5; nd=1. 5 Случай трех замен приводит к 6 возможным путям по 3 шага. Оценка скоростей по формуле Джукса-Кантора. доли син. /несин. замен Вместо p необходимо подставить соотв. ps или pn, получим ds или dn
Подсчет числа замен. Случай одной замены: GUU(Val) -> GUA (Val) sd=1; nd=0. Случай двух замен: Рассматриваются несколько возможных путей, которые считаются равновероятными. UUU(Phe) -> GUA(Val) : (1): UUU(Phe) -> GUU(Val) -> GUA(Val) sd=1; nd=1. (2): UUU(Phe) -> UUA(Leu) -> GUA(Val) sd=0; nd=2. (1)+(2): sd=0. 5; nd=1. 5 Случай трех замен приводит к 6 возможным путям по 3 шага. Оценка скоростей по формуле Джукса-Кантора. доли син. /несин. замен Вместо p необходимо подставить соотв. ps или pn, получим ds или dn
 Оценка отношения для ветвей филогенетического дерева Оценка эволюционных расстояний для пар последовательностей дерева двух типов (dn, ds) Оценка длин для ветвей дерева двух типов (dn, ds) призаданной топологии дерева. Пример: анализ белка катионного белка (eosinophil cationic protein ECP) и нейротоксинов эозинофилов (eosinophil neurotoxin EDN) генов в приматах после дупликации (J. Zhang et al, (1998) PNAS v. 95 p 3708). ECP EDN
Оценка отношения для ветвей филогенетического дерева Оценка эволюционных расстояний для пар последовательностей дерева двух типов (dn, ds) Оценка длин для ветвей дерева двух типов (dn, ds) призаданной топологии дерева. Пример: анализ белка катионного белка (eosinophil cationic protein ECP) и нейротоксинов эозинофилов (eosinophil neurotoxin EDN) генов в приматах после дупликации (J. Zhang et al, (1998) PNAS v. 95 p 3708). ECP EDN
 Анализ в участках последовательностей белков и в отдельных позициях Методы эволюционных расстояний. 1. Использование сканирующего окны (простой но неточный) 2. Реконструирование предковых последовательностей и подсчет замен в каждом кодоне (Fitch et al. 1997; Suzuki & Gojobori 1999): TTC T A TTC C T TTC ATC C A 3 несинонимических замены 1 синонимическая замена TTA T A TAT TTT Методы максимального правдоподобия (PAML) TTT
Анализ в участках последовательностей белков и в отдельных позициях Методы эволюционных расстояний. 1. Использование сканирующего окны (простой но неточный) 2. Реконструирование предковых последовательностей и подсчет замен в каждом кодоне (Fitch et al. 1997; Suzuki & Gojobori 1999): TTC T A TTC C T TTC ATC C A 3 несинонимических замены 1 синонимическая замена TTA T A TAT TTT Методы максимального правдоподобия (PAML) TTT
 ввел матрицу замен") Пакет программ PAML Для описания эволюции на уровне кодонов Янг (1994) ввел матрицу замен кодонов 61 х61 Основные параметры: Отношение скоростей транзиций – трансверсий Частоты встречаемости кодонов I Отношение скоростей несинонимических и синонимических замен =dn/ds
Пакет программ PAML Для описания эволюции на уровне кодонов Янг (1994) ввел матрицу замен кодонов 61 х61 Основные параметры: Отношение скоростей транзиций – трансверсий Частоты встречаемости кодонов I Отношение скоростей несинонимических и синонимических замен =dn/ds
 Значение элементов матрицы замен кодонов • Метод реализован в пакете программ PAML и позволяет выявлять адаптивный режим эволюции.
Значение элементов матрицы замен кодонов • Метод реализован в пакете программ PAML и позволяет выявлять адаптивный режим эволюции.
 25 позиций белка") Анализ замен в отдельных позициях (MHC, метод: PAML, Yang et al) 25 позиций белка с адаптивным режимом Эволюции располагались в основном в районе связывания с эпитопом
Анализ замен в отдельных позициях (MHC, метод: PAML, Yang et al) 25 позиций белка с адаптивным режимом Эволюции располагались в основном в районе связывания с эпитопом
 Проверка адаптивного режима эволюции для ветвей филогенетического дерева
Проверка адаптивного режима эволюции для ветвей филогенетического дерева
 Задачи: адаптивная эволюция белков в геномах Постановка задачи. Определить по каким генам в геноме шла адаптивная эволюция. Возможные кандидаты: гены иммунной системы или защиты; у вирусов – гены оболочки (уход от иммунной системы хозяина); гены размножения.
Задачи: адаптивная эволюция белков в геномах Постановка задачи. Определить по каким генам в геноме шла адаптивная эволюция. Возможные кандидаты: гены иммунной системы или защиты; у вирусов – гены оболочки (уход от иммунной системы хозяина); гены размножения.
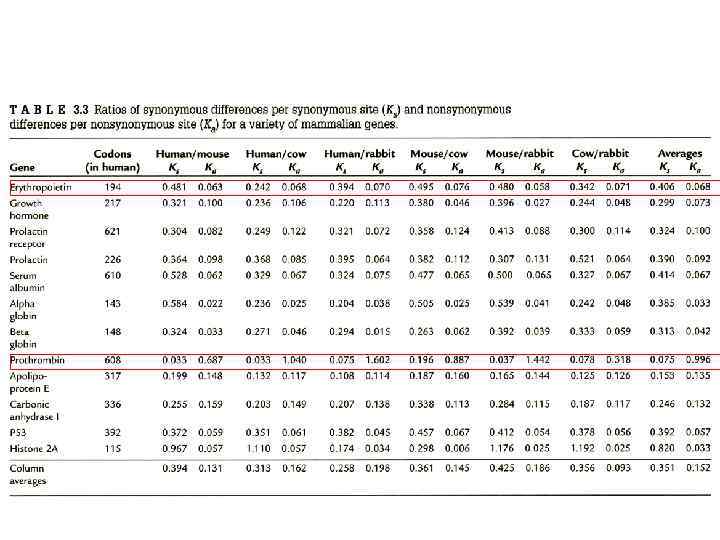
 Сравнение скоростей синонимических/несинонимических замен Оценки скоростей нуклеотидных замен на 1 позицию на 1000 млн. Лет при сравнении некоторых белков человека и грызунов (время дивергенции ~80 млн. лет согласно ископаемым) Ген Число кодонов histone 3 135 K(несин. ) K(син. ) 0. 00 6. 38 actin beta 349 0. 03 3. 13 myoglobin 153 0. 56 4. 44 In-gamma 136 2. 79 8. 59
Сравнение скоростей синонимических/несинонимических замен Оценки скоростей нуклеотидных замен на 1 позицию на 1000 млн. Лет при сравнении некоторых белков человека и грызунов (время дивергенции ~80 млн. лет согласно ископаемым) Ген Число кодонов histone 3 135 K(несин. ) K(син. ) 0. 00 6. 38 actin beta 349 0. 03 3. 13 myoglobin 153 0. 56 4. 44 In-gamma 136 2. 79 8. 59
 Сравнительная эволюция белков в геномах мыши, шимпанзе и человека
Сравнительная эволюция белков в геномах мыши, шимпанзе и человека
") 7645 генов шимпанзе сравнивались с ортологами мыши и человека Использовалась программа PAML (подпрограмма codeml)
7645 генов шимпанзе сравнивались с ортологами мыши и человека Использовалась программа PAML (подпрограмма codeml)
 Проверялись две модели эволюции: • М 1 Замены на ветке к человеку являются нейтральными. (отвергнута для 1216 генов (15. 9%) при p<0. 05) • М 2 Замены в линии человека либо нейтральны, либо дестабилизирующие (альтернатива – наличие адаптивных замен) (отвергнута для 667 генов (8. 7%) при p<0. 05) Ч M ~5 -8 млн лет назад Ш
Проверялись две модели эволюции: • М 1 Замены на ветке к человеку являются нейтральными. (отвергнута для 1216 генов (15. 9%) при p<0. 05) • М 2 Замены в линии человека либо нейтральны, либо дестабилизирующие (альтернатива – наличие адаптивных замен) (отвергнута для 667 генов (8. 7%) при p<0. 05) Ч M ~5 -8 млн лет назад Ш
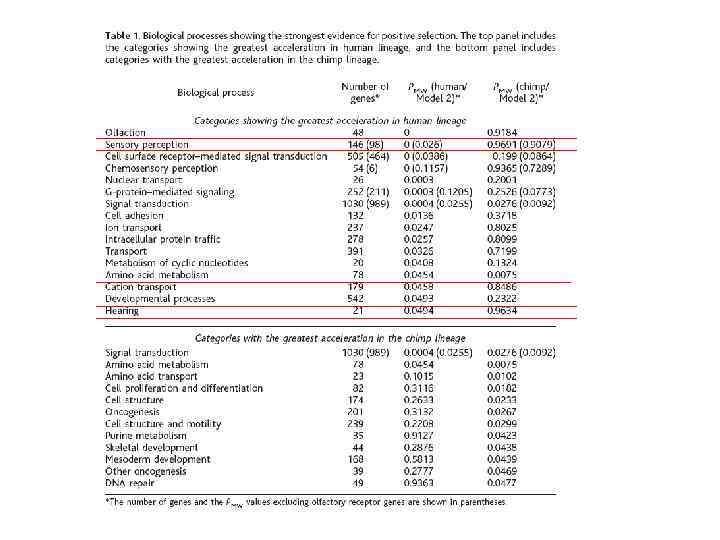
 • Для человека характерно быстрое") Обоняние • Обоняние определяется рецепторами обоняния (olfactory receptors OR) • Для человека характерно быстрое образование псевдогенов для этих рецепторов, потеря их функции. Этот процесс представляется как быстрое накопление несинонимичных замен.
Обоняние • Обоняние определяется рецепторами обоняния (olfactory receptors OR) • Для человека характерно быстрое образование псевдогенов для этих рецепторов, потеря их функции. Этот процесс представляется как быстрое накопление несинонимичных замен.
 • Гены, вовлеченные в метаболизм аминокислот так же эволюционировали адаптивно • Причина – разница в питании, в частности человек привык есть мясо (белки)
• Гены, вовлеченные в метаболизм аминокислот так же эволюционировали адаптивно • Причина – разница в питании, в частности человек привык есть мясо (белки)
 Гены развития человека • Два основных класса этих генов: – Развитие скелета – Нейрогенез (развитие интеллекта) • Часть генов связаны с развитием беременности – Progesterone receptor (PGR)
Гены развития человека • Два основных класса этих генов: – Развитие скелета – Нейрогенез (развитие интеллекта) • Часть генов связаны с развитием беременности – Progesterone receptor (PGR)
 Речь и слух • Некоторые гены в этих системах у человека эволюционировали адаптивно – Forkhead-box P 2 transcriptions factor, which is involved in speech development – Alpha tectorin, protein plays major role in the tectorial membrane of the inner ear
Речь и слух • Некоторые гены в этих системах у человека эволюционировали адаптивно – Forkhead-box P 2 transcriptions factor, which is involved in speech development – Alpha tectorin, protein plays major role in the tectorial membrane of the inner ear
? Слабее различать запахи, способность расщеплять аминокислоты") Что делает нас людьми (по сравнению с обезьянами)? Слабее различать запахи, способность расщеплять аминокислоты (есть мясо), слышать, продвинутый нейрогенез
Что делает нас людьми (по сравнению с обезьянами)? Слабее различать запахи, способность расщеплять аминокислоты (есть мясо), слышать, продвинутый нейрогенез
 Вопросы к экзамену Что такое эволюционное расстояние? В чем отличие модели нуклеотидных замен Кимуры от модели Джукса-Кантора? Классификация аминокислотных замен в белках. Особенности модели эволюции аминокислотных последовательностей Дайхоф. Что такое мутабильность аминокислоты? Каким образом матрицы сходства аминокислот (матрица весов сравнения аминокислот) связаны с матрицами скоростей замен? Что такое филогенетическое дерево? Какие существуют методы построения филогенетических деревьев? Метод расстояний на примере UPGMA и его недостатки.
Вопросы к экзамену Что такое эволюционное расстояние? В чем отличие модели нуклеотидных замен Кимуры от модели Джукса-Кантора? Классификация аминокислотных замен в белках. Особенности модели эволюции аминокислотных последовательностей Дайхоф. Что такое мутабильность аминокислоты? Каким образом матрицы сходства аминокислот (матрица весов сравнения аминокислот) связаны с матрицами скоростей замен? Что такое филогенетическое дерево? Какие существуют методы построения филогенетических деревьев? Метод расстояний на примере UPGMA и его недостатки.
 Вопросы к экзамену Какие существуют режимы отбора? Чем характерна фиксация нейтральных замен? Каково соотношение по приспособленности мутаций в белках согласно Кимуре? Что такое гипотеза молекулярных часов? Что такое синонимические и несинонимические замены? Каким образом оценить тип отбора для двух выровненных последовательностей?
Вопросы к экзамену Какие существуют режимы отбора? Чем характерна фиксация нейтральных замен? Каково соотношение по приспособленности мутаций в белках согласно Кимуре? Что такое гипотеза молекулярных часов? Что такое синонимические и несинонимические замены? Каким образом оценить тип отбора для двух выровненных последовательностей?