Генные болезни Дивинская.ppt
- Количество слайдов: 132



Фенилкетонурия, муковисцидоз, целиакия, ГЕННЫЕ БОЛЕЗНИ

Генные болезни Обусловленные мутациями в генах наследуются по законам Менделя: 25% больных 50 % носителей 25 % здоровых

Виды генных мутаций: Миссенс - замена нуклеотида в кодирующей части гена Нонсенс – замена нуклеотида в кодирующей части гена, образование стоп-кодона Инсерция - вставка отрезка ДНК размерами от одного нуклеотида до нескольких генов

Виды генных мутаций: Точечная мутация – изменение одной пары оснований Сплайсинг -вырезание интронов и объединение экзонов в зрелую м. РНК Экспансия – увеличение числа копий тандемных последовательностей

Виды генных мутаций: Делеция – утрата сегмента ДНК размером от одного до неск. генов Инверсия – поворот на 180 сегмента ДНК размером от 2 -х нуклеотидов до нескольких генов Дупликация – удвоение сегмента ДНК размером от одного нуклеотида до нескольких генов

Классификация генных болезней I. Генетический принцип: - Аутосомно-доминантные Аутосомно-рецессивные Х-сцепленные доминантные Х-сцепленные рецессивные У-сцепленные Митохондриальные

II. Клинический принцип: Пихические Нервные Нервно-мышечные Кожные Глазные Болезни опорно-двигательного аппарата Болезни крови Эндокринные Болезни сердечно-сосудистой системы Болезни мочеполовой системы Болезни желудочно-кишечного тракта Болезни легких
 Болезни ОБВВ: Углеводного Аминокислотного Липидного Обмена витаминов")
III. Патогенетическая классификация - ВПР (моногенной природы) Болезни ОБВВ: Углеводного Аминокислотного Липидного Обмена витаминов Обмена металлов Комбинированные состояния

Нарушение обмена аминокислот МЕТАБОЛИЧЕСКИЕ БОЛЕЗНИ
 ФКУ – врожденная поломка аминокислотного обмена (1934 г. -")
ФЕНИЛКЕТОНУРИЯ – ФКУ (фенилпировиноградная олигофрения) ФКУ – врожденная поломка аминокислотного обмена (1934 г. - А. Феллинг). Ген локализован на длинном плече 12 q хромосомы (12 q 22 -24), содержит 70000 пар нуклеотидов, наиболее частая (мажорная) мутация R 408 W Известно около 300 мутаций гена.

Частота ФКУ Европейские страны - 1 : 10. 000 Турция – 1 : 2600 Китай - 1 : 16. 000 Швеция – 1 : 30. 000 Ирландия - 1 : 4500 Белоруссия - 1 : 6000 Япония – 1 : 119. 000 Крым – 1 : 7355

Патогенетические аспекты ФКУ Недостаточность фермента Ф-А-гидроксилазы Нарушение гидроксилирования Ф-А в тирозин (из тирозина образуются тироксин, меланин, адреналин) Накопление Ф-А в крови (фенилаланинемия)
 с мочой Нарушение миелинизации аксонов в ЦНС")
Выделение фенилпировиноградной и фенилуксусной к-т (кетокислот) с мочой Нарушение миелинизации аксонов в ЦНС Снижение меланина - гипопигментация кожи, волос, радужки Снижение адреналина – артериальная гипотония
 форма 2. Атипичные формы: - стойкая гипер-фенилаланинемия -")
Клинические формы ФКУ: 1. Типичная (классическая) форма 2. Атипичные формы: - стойкая гипер-фенилаланинемия - транзиторная гипер-фенилаланинемия - материнская ФКУ

Классическая форма: Дети рождаются здоровыми, доношенными При поступлении Ф-А с грудным молоком: ▪ повышенная возбудимость ▪ гиперрефлексия (сухожильных) ▪ гипертонус мышц ▪ тремор, судорожные (эпи-) припадки ▪ реже гиперкинезы или парезы ▪ «мышиный» запах
")
К 4 -5 месяцу развиваются: ЗПР прогрессирует : (к 4 годам дебильность, имбицильность, идиотия) сонливость, вялость не фиксирует взгляд не интересуется игрушками Рвота, диспепсия
 голубые глаза задержка физического развития задержка")
Клинические симптомы светлеют волосы и кожа (недостаток меланина) голубые глаза задержка физического развития задержка прорезывания зубов задержка речи артериальная гипотония (недостаток адреналина)

Нейросонография : внутричерепная гипертензия ЭЭГ: пароксизмальная активность МРТ: изменения в белом веществе, атрофия коры ГМ
 Отсутствуют клинические проявления Нормальное")
Атипичные формы: Стойкая ГФА: Незначительное повышение Ф-А (15 -20 %) Отсутствуют клинические проявления Нормальное развитие без соблюдения специфической элиминационной диеты
 После дозревания Ф-А-трансферазы нормализуются")
Атипичные формы: Транзиторная ГФА: Умеренное повышение Ф-А (чаще у недоношенных) После дозревания Ф-А-трансферазы нормализуются уровни Ф-А и тирозина

Атипичные формы: Материнская ФКУ: - Беременная с ФКУ Накопление Ф-А в плаценте Проникновение Ф-А через ГЭБ Рождение ребенка с УО

Диагностика ФКУ: Проба Фелинга : (2 -5 мл свежей мочи + 6 -10 капель 10% хлорида железа – Fe. Cl 3. При наличии Ф-А моча окрасится в сине-зеленый цвет. Тест Гатри: на диск фильтровальной бумаги капнуть кровь ребенка (не ранее 4 дня жизни, чтобы накопился Ф-А при употреблении грудного молока).

Диагностические критерии ФКУ Ф-А крови выше 10 -15 мг% (норма Ф-А крови до 2 мг%) Ф-А мочи выше 100 мг% Флюороскан – точный метод компьютерной диагностики ДНК-диагностика

Лечение ФКУ Строгая диета до 4 летнего возраста (под контролем Ф-А крови – оптимальная концентрация Ф-А у больных 3 -8 мг%) Исключить продукты, содержащие Ф-А (мясо, молоко, яйца) Включить специальные смеси без Ф-А (или с минимальным его содержанием Ф-А): Лофенолак Фенил-Фри Тетрафен Минафен Фенил-дон

Муковисцидоз - наследственное аутосомно-рецессивное мультисистемное заболевание, развивающееся вследствие продукции экзокринными железами секрета повышенной вязкости, с развитием вторичных изменений в бронхо-легочной, пищеварительной и репродуктивной системах
. 1")
Частота встречаемости МВ Самое частое наследственное з-е: (в 2 раза чаще, чем ФКУ). 1 : 2500 1 : 3000 Среди африканцев 1 : 100. 000. Частота гетерозигот в Европе высока до 5% населения. Аутосомно-рецессивный тип наследования.
 состоит из")
ПАТОГЕНЕЗ: Ген МВ расположен на 7 хромосоме (7 q 31 – 32) состоит из 250000 пар оснований. Экспрессия гена ограничена г. о. эпителиальными клетками. В основе патогенеза лежит нарушение транспорта ионов хлора и натрия через клеточные мембраны. Обнаружено около 500 мутаций. Наиболее часто (70%) – делеция 3 пар оснований в 508 положении ( F 508) полипептидной цепи.
.")
Патогенез МВ: Выводные протоки поджелудочной железы закупориваются, слизь не выводится, образуются кисты (кистозный фиброз). Ферменты поджелудочной железы не поступают в просвет кишечника (внешнесекреторная недостаточность). Гиперсекреция бронхиального секрета (повышенной вязкости) приводит к обтурации мелких бронхов и ателектазам.

Патогенез МВ: Подобные процессы развиваются в придаточных пазухах, в канальцах семенников В потовой жидкости повышена концентрация ионов натрия и хлора (основной диагностический признак)
 С преимущественным поражением поджелудочной железы Без преимущественного поражения")
Классификация МВ: Мекониальный илеус (врожденная форма) С преимущественным поражением поджелудочной железы Без преимущественного поражения поджелудочной железы С поражением репродуктивной функции
: избыточное количество")
Клиническая картина Мекониальный илеус – врожденная форма МВ (1% от всех форм): избыточное количество густого мекония в кишечнике. Симптомы кишечной непроходимости: - Рвота с примесью желчи - Увеличение живота - Отсутствие мекония - Симптомы перитонита - Оперативное лечение.
 Пг:")
С преимущественным поражением поджелудочной железы Кишечная форма: (5 -10% от всех форм МВ) Пг: недостаточность панкреатических ферментов (липазы): КЛИНИКА: Гипотрофия, «симптом соленого ребенка» , Полифекалия: светлый, зловонный, замазкообразный с большим количеством жира Живот вздут, жировая инфильтрация печени, холестатический гепатит, цирроз.
")
Копроцитология: Стеаторея, жирные кислоты Креаторея Клетчатка Зерна крахмала Иодоформные бактерии (брожение)
: Симптомы обструкции на фоне")
Без преимущественного поражения поджелудочной железы Бронхолегочная форма: (15 -20% ): Симптомы обструкции на фоне ОРВИ Присоединение инфекции- гнойнообструктивный бронхит, тяжелые пневмонии (рецидивирующего характера). Пневмонии двусторонние!!! (Двусторонние при гематогенном сепсисе). Пневмония в период новорожденности – подозрение на МВ.

Без преимущественного поражения поджелудочной железы Флора: синегнойная палочка, золотистый стафилококк, гемофильная палочка или ассоциации – устойчива к а/б. Осложения: бронхоэктазы, ателектазы, эмфизема, пневмосклероз, легочное сердце. Рентген: «волосатое» сердце (усиленный легочный рисунок). Умирают от ДН или от сердечной недостаточности
 трехкратно с интервалом 3 дня")
Диагностика МВ: Потовая проба (биохимический анализ Na и Cl) трехкратно с интервалом 3 дня (для МВ все три д. б. + ) На спину лакмусовую бумажку, закутать и горячее питье на 30 минут (предварительно гигиеническая ванна). Норма хлоридов: до 1 года – 40 ммоль/л старше 1 года – 60 ммоль/л Подозрение на МВ – хлориды - выше 60 ммоль/л Диагностически значимый уровень: хлоридов – выше 100 ммоль/л натрия - выше 70 ммоль/л
. 6 пробирок по 10 мл")
ПРОБА Швахмана Проба Швахмана – рентген-пленочный тест (желатиновый тест). 6 пробирок по 10 мл физ. р-ра ( в первую пробирку + фекалии) и переносят по 1 мл из пробирки в пробирку – получают разведения. на засвеченную рентген-пленку капают по капле, на 1 час в термостат. наличие просветлений (д. б. во всех 6 полях), т. е. протеолитическая активность трипсина не снижена. Норма – в разведении 1 : 320 Снижена активность ферментов трипсина кишечного содержимого 1 : 60 1 : 80 МВ: 1 : 10 1 : 20 или везде отсутствует

Диагностика МВ: Ногтевой тест : в соскобе ногтя хлориды и натрий Пилокарпиновый тест (хлориды пота) Пренатальная ДНК-диагностика МВ для большинства мутаций (ПЦР). УЗИ гепатобилиарной системы и панкреас Спирография и рентгенография грудной клетки Копроцитограмма

Лечение МВ Ферменты: панцитрат, мезим, мексаза, фестал, креон, панзинорм, дигестал Муколитики: ацетилцистеин, цитохром – С (на тканевое дыхание). Антибиотики в период обострения Витамины А, Е! эссенциале – постоянно (стабилизаторы клеточных мембран, противовоспалительное д-е) Бронхосанация

ЦЕЛИАКИЯ
 Синонимыглютеновая энтеропатия, глютеновая болезнь, нетропическая спру, болезнь Ги-Гертера-Гюбнера")
ЦЕЛИАКИЯ (coeliakia, coeliac disease) Синонимыглютеновая энтеропатия, глютеновая болезнь, нетропическая спру, болезнь Ги-Гертера-Гюбнера

Страна Клинические случаи USA 1: 10 000 1: 111 1: 4. 500 1: 198 1: 2. 300 1: 500 Италия Финляндия Бразилия 1: 400 Аргентина 1: 167 Украина ? Серологическая диагностика ?
 ЦЕЛИАКИЯ – хроническое,")
Формулировка диагноза (Передерий В. Г. с соавт. , 2005 г. ) ЦЕЛИАКИЯ – хроническое, генетически обусловленное иммунно-опосредованное заболевание, которое характеризуется стойкой непереносимостью продуктов, которые содержат (пшеницу, рожь, ячмень, овес), поражает слизистую оболочку тонкого кишечника и ведет к развитию ее атрофии, проявляется синдромом мальабсорбции и может иметь полностью обратимое течение за счет восстановления структуры слизистой оболочки после полного исключения из пищевого рациона глютена.
 2. Наследственная")
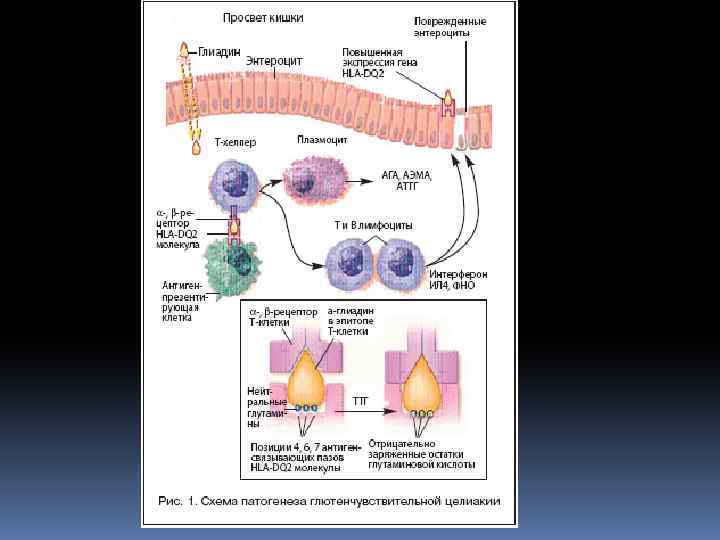
ЭТИОПАТОГЕНЕЗ ЦЕЛИАКИИ Для развития заболевания нужны два условия: 1. Белок злаковых ((глютен) 2. Наследственная предрасположенность Целиакия наследуется по аутосомно-доминантному типу и является полигенным заболеванием. Существует ассоциация целиакии с локусами гена главного комплекса гистосовместимости, расположенного на 6 хромосоме. У 92 -95 % больных целиакией определяется генетическая предрасположенность по системе HLA (DQ 2 / DQ 8)
 латентная")
КЛАССИФИКАЦИЯ ЦЕЛИАКИИ Формы заболевания: Степень тяжести: . Периоды заболевания: типичная атипичная (скрытая, немая) латентная легкая средне-тяжёлая обострения ремиссии Пример диагноза: Целиакия, типичная форма, средне-тяжёлая степень тяжести, период обострения, с наличием синдрома мальабсорбции, железодефицитной анемией, с отставанием в массе и росте.

ТИПИЧНАЯ форма целиакии Дебютирует: в раннем детском возрасте, в среднем на 3 -12 мес. жизни. Первые признаки: появляются спустя несколько месяцев после введения в рацион ребенка продуктов, которые содержат злаки (каши). диспептические явления, которые расцениваются как проявления незрелости пищеварительного канала ребенка в данном возрасте. Постепенно состояние ребенка ухудшается, нарастают симптомы, появляется задержка роста и массы. Морфологические признаки: разной степени выраженности.
 форма целиакии Характеризуется : 1) отсутствием типичных клинических проявлений 2) наличием")
АТИПИЧНАЯ (скрытая, немая) форма целиакии Характеризуется : 1) отсутствием типичных клинических проявлений 2) наличием специфических морфологических признаков поражения слизистой оболочки тонкого кишечника, что объясняется сегментарным поражением тонкой кишки, когда неповрежденные участки кишки полностью компенсируют его функцию Манифестация заболевания возможна: 1) под влиянием тригерных факторов 2) при увеличении глютеновой нагрузки
 отсутствие клинических проявлений 2) отсутствие")
ЛАТЕНТНАЯ форма целиакии Присуща: «генетически скомпрометированным» пациентам. Характерно: 1) отсутствие клинических проявлений 2) отсутствие морфологических изменений Реализация генетических дефектов возможна при: 1) влиянии тригерных факторов 2) увеличении глютеновой нагрузки Латентную форму предболезнь можно рассматривать как

ТЕЧЕНИЕ ЦЕЛИАКИИ Характеризуется: чередованием периодов обострения и ремиссии. Продолжительность самостоятельной ремиссии: - от нескольких месяцев - до нескольких лет Обострение протекает: с прогрессирующим поражением органов и систем.

Наиболее характерные признаки современной «типичной» целиакии 1. Неопределенная боль в брюшной полости и/или ощущение дискомфорта. 2. Вздутие живота, чувство переполнения в брюшной полости. 3. Сниженный аппетит. 4. Диарея. 5. Полифекалия. 6. Зловонный стул. 7. Потеря массы.

Наиболее распространённые внекишечные проявления целиакии 1. Чувство жара и боли в ротовой полости (афтозный стоматит). 2. Железо-фолиеводефицитная анемия. 3. Аллергические реакции. 4. Герпетиформный дерматит Дюринга. 5. Аутоиммунные заболевания (сахарный диабет 1 типа, тиреотоксикоз, болезнь Аддисона и др. ). 6. Ревматоидные и серонегативные артриты. 7. Лимфомы. 8. Кишечные кровотечения.

ДИАГНОСТИКА ЦЕЛИАКИИ 1. Клинические проявления. 2. Фиброгастродуоденоскопия постбульбарного отдела 12 -перстной кишки с прицельной биопсией. 3. Морфологический субстрат – является «золотым» стандартом диагностики. 4. Капсульная эндоскопия. 5. Серологическая диагностика.


Marsh 2 Очаговая форма Гиперпластическая стадия Marsh 2

СЕРОЛОГИЧЕСКАЯ ДИАГНОСТИКА В качестве серологических маркёров используются: 1. Антитела к глиадину – антиглиадиновые антитела (АГА Ig A, Ig M) 2. Антитела к компонентам соединительной ткани: • эндомизию – антиэндомизиальные антитела (АЭМА) • ретикулину – антиретикулиновые антитела (АРА) 3. Антитела к ткани тощей кишки и тканевой трансглутаминазе (т. ТГ, t. TG - т. ТГА)

Серологические маркёры, их специфичность и чувствительность Антитела Чувствительность, % Специфичность, % АГА Ig A 98 % дети 86 % дети АГА Ig А 75 -90 % взрослые 82 -95 % взрослые АГА Ig G 89 % дети 95, 5 % дети АГА Ig. G 69 -84 % взрослые 73 -90 % взрослые АЭМА Ig A 85 -98 % 98 -99 % АРА Ig A 68 -91 % 99 -100 % т. ТГА Ig A 95 -98 % 94 -98 %

Строгая безглютеновая диета – является базисной терапией при целиакии Врач должен проинформировать не только больного, но и его родных на чём базируется лечение: 1) необходимо чётко придерживаться диеты 2) соблюдать диету на протяжении всей жизни, а не только в период обострения 3) ознакомить с перечнем разрешённых и запрещенных продуктов 4) медикаментозная терапия не даёт избежать безглютеновой диеты; 5) употребление даже 0, 5 г глютена в сутки значительно снижает вероятность улучшения состояния, а доза 1 -2 г в сутки считается токсичной для больного;

Продукты, противопоказанные больным целиакией Злаки : - - пшеничная и ржаная мука выпечка из пшеничной и ржаной муки манная, овсяная, ячменная и перловая каши пшеничные, овсяные и ячменные хлопья Мясо : - фарш, приготовленный в магазине или на производстве - колбаса вареная и копчёная - сосиски, сардельки - ветчина - любые полуфабрикаты - мясные консервы Рыба : - рыбные консервы, особенно в томатном соусе

продолжение: Молочные продукты: - молоко порошковое - молоко шоколадное - йогурт - глазурованные сырки - майонезы Сладости : - карамель с начинкою - шоколадные конфеты с начинкою - шоколад молочный - любые конфеты и шоколад, которые содержат солод или крахмал - восточные сладости - повидло - мороженное
 - напитки с кофе, ячменём,")
продолжение: Напитки : - соки в пакетах (детского питания) - напитки с кофе, ячменём, какао - пиво - водка пшеничная Приправы : - кетчупы и томатные пасты - многокомпонентные приправы, которые содержат солод и пшеничную муку Овощи и фрукты : без ограничений Жиры : без ограничений.
 продукты путём питания, приготовленные промышленным 2) пищевые добавки, стабилизаторы, красители,")
СКРЫТЫЕ ИСТОЧНИКИ ГЛЮТЕНА 1) продукты путём питания, приготовленные промышленным 2) пищевые добавки, стабилизаторы, красители, эмульгаторы, некоторые медикаменты (капсулы) 3) компоненты поливитаминных комплексов 4) зубные пасты 5) жевательные резинки Глютен может попадать на стол путём банального загрязнения продуктов на прилавках магазинов, в тостерах, в микстерах и любой кухонной посуде.

Наследственные болезни углеводного обмена Галактоземия Фруктоземия Алактазия Мальтазная недостаточность Сахаридазная недостаточность Гликогенозы

Галактоземия Наследственная энзимопатия, обусловленная нарушением метаболизма галактозы в связи с дефицитом фермента галактозо-1 -фосфат-уридилтрансферазы. Аутосомно-рецессивный тип наследования 1: 30. 000 1: 70. 000

Патогенез галактоземии: П/г: в результате отсутствия фермента в тканях организма накапливается избыточное количество продуктов неполного распада галактозы: Гипогликемия Ацидоз Гипокалиемия

Клиническая симптоматика: Манифестация сразу после рождения, когда ребенок начинает получать молоко: Рвота неукротимая Диарея Гипотрофия Желтуха Мышечная гипотония Арефлексия в т. ч. и рефлексы НВ Задержка ПМР и позже УО Гепатомегалия
,")
Клиническая симптоматика: Геморрагии на коже и кровотечения Нейротоксикоз, судороги, глазодвигательные расстройства (косоглазие и нистагм), затруднение сосания и глотания Лицо гипомимично, отрицательный эмоциональный комплекс Катаракта Цирроз печени, асцит

Диагностика галактоземии: Уровень галактозы в крови с помощью ТСХ на бумаге Галактоза в моче Активность галактозо-1 -фосфат-уридилтрансферазы в эритроцитах Патоморфология: жировая дистрофия печени, цирроз, катаракта, дегенерация корковых нейронов, атрофия и склероз мозгового вещества. При тяжелой форме м. б. летальный исход

Лечение галактоземии Лечение: исключить из рациона молоко и все продукты с галактозой. Смеси «Галактомин» , «Нутрилак»

Гомоцистинурия Дефект фермента: циста-тионин- синтетазы Гомоцистин не превращается в метионин пг: неполноценность соединительной ткани

Фенотипические проявления: Туловище укорочено Конечности удлинены Воронкообразная грудная клетка Крыловидные лопатки Башенный череп Светлые волосы Голубые глаза Подвывих хрусталика Пролапс митрального клапана

Кожа: сухость, складчатость, депигментированные пятна, телеангиэктазии Тромбоэмболии: (гомоцистин оседает на интиме сосудов, активизируется XII фактор) Диагностика: микробиологический метод : микробы размножаются в присутствии гомоцистина) Лечение: Диета с ограничением белка (метионина) Витамин В 6

Врожденный гипотиреоз КЛИНИЧЕСКИЕ МАРКЕРЫ Беременность более 40 недель Масса тела при рождении более 3500 г Женский пол (девочки : мальчики = 2 : 1) Позднее отхождение мекония (более 24 часов) Поздно отпадает пуповинный остаток Длительное заживление пупочной ранки Затянувшаяся желтуха (более 3 недель) Мышечная гипотония, пупочная грыжа

КЛИНИЧЕСКИЕ МАРКЕРЫ Отечность лица, тыльных поверхностей кистей, стоп, надключичных ямок Грубый, низкий тембр голоса Полуоткрытый рот, увеличенный язык (макроглоссия) и губы Шелушение, сухость, бледность, «мраморность» кожи Редко плачет, не реагирует на голод, на какойлибо дискомфорт Гипотермия Открытый малый родничок Слабый сосательный и глотательный рефлексы

ДЕТИ ГРУДНОГО И БОЛЕЕ СТАРШЕГО ВОЗРАСТА Сниженный аппетит, недостаточная прибавка массы тела Запоры, метеоризм (увеличенный живот) Пупочная грыжа Мышечная гипотония Задержка психомоторного развития (в последующем умственная отсталость – кретинизм)
 Задержка появления ядер окостенения (отставание костного возраста от")
Врожденный гипотиреоз Задержка роста (тиреогенный нанизм) Задержка появления ядер окостенения (отставание костного возраста от паспортного) Позднее прорезывание зубов и нарушение порядка их появления Гипотермия (плохая переносимость холода) Позднее закрытие родничков (к 8 -9 годам жизни) Эмоциональная сглаженность (безразличный взгляд, редкий плач, отсутствие реакции на речь, звук, предметы), сонливость, судорожные подергивания, стереотипность движений головой

Врожденный гипотиреоз

Врожденный гипотиреоз Характерные черты телосложения: короткие конечности, широкие стопы и кисти, относительно большая голова, поясничный лордоз, ноги полусогнуты (раскачивающаяся походка) Волосы тусклые, грубые, ломкие, замедленный рост, диффузная алопеция Ногти с трещинами, практически не растут Широкая переносица, рот открыт, язык распластан, отпечатки зубов
")
Врожденный гипотиреоз Брадикардия, снижение АД, расширение границ, приглушенность тонов сердца (снижение вольтажа на ЭКГ) У более старших детей – атаксия, спастическая диплегия, нейросенсорная тугоухость, косоглазие, замедленная речь, неловкость, замедленность движений, задержка полового развития Увеличение печени, снижение кислотности желудочного сока Нормохромная, нормоцитарная анемия, гиперхолестеринемия, повышение ТТГ, снижение Т 4.

СКРИНИНГ ВРОЖДЕННОГО ГИПОТИРЕЗА Забор крови у доношенных новорожденных производится на 4 -5 день жизни (у недоношенных – на 7 -14 день). Трактовка результатов: ТТГ до 20 м. ЕД/л – вариант нормы
. Критерии эффективности лечения: Отсутствие клинических симптомов")
ЛЕЧЕНИЕ ГИПОТИРЕОЗА Препаратом выбора является левотироксин (α-тироксин, эутирокс). Критерии эффективности лечения: Отсутствие клинических симптомов Нормальные темпы роста и развития ТТГ: 0, 2 – 2, 0 м. ЕД/л
 Частота АГС - 1 : 10000 (13 всех")
Адреногенитальный синдром (ВРОЖДЕННАЯ ГИПЕРПЛАЗИЯ КОРЫ НАДПОЧЕЧНИКОВ) Частота АГС - 1 : 10000 (13 всех случаев гермафродитизма) АГС - группа наследственных заболеваний, в основе которых лежат ферментативные дефекты адреналового стероидогенеза Все формы АГС наследуются по аутосомнорецессивному типу.
 1. Врожденная (классическая)")
Клинические формы АГС I. Простая вирильная форма (частичный дефицит 21 -гидроксилазы) 1. Врожденная (классическая) 2. Периода пубертата (неклассическая) II. Сольтеряющая форма (выраженный дефицит 21 -гидроксилазы) III. Гипертоническая форма (дефицит 11 -гидроксилазы)
: Вирильный синдром – это симптомокомплекс, развивающийся")
ПРОСТАЯ ВИРИЛЬНАЯ ФОРМА Простая вирильная форма врожденная (классическая): Вирильный синдром – это симптомокомплекс, развивающийся в женском организме вследствие избытка андрогенов.
. Кортизол – низкий")
Патогенез : П/Г: дефект биосинтеза кортизола стимулирует продукцию адренокортикотропного гормона (АКТГ). Кортизол – низкий уровень АКТГ – высокий уровень эти изменения приводят к гиперплазии коры надпочечников Накапливается 17 -гидрокси - прогестерон (увеличен в 10 и более раз) Это предшественник кортизола, который превращается в андрогены.

Вирильная форма У девочек в период внутриутробного развития идет вирилизация наружных гениталий (в результате избытка андрогенов), нарушается половая дифференцировка (т. н. интерсексуализм). Т. Е. неопределенное строение наружных гениталий затрудняет определение половой принадлежности ( КАРИОТИП !). Если кариотип 46 /ХХ – это вирильный синдром т. е. ( ложный женский гермафродитизм).

Вирильная форма Наружные гениталии сформированы по гетеросексуальному типу (клитор гипертрофирован, большие половые губы напоминают мошонку, вагина и уретра открываются единым мочеполовым отверстием (урогенитальным синусом), расположенным либо на промежности, либо на стволе гипертрофированного клитора). Для оценки степени вирилизации (5 степеней) - шкала ПРАДЕРА В дальнейшем у девочек не растут молочные железы, не наступают менструации

Вирильный синдром

Вирильная форма У новорожденных мальчиков явных нарушений не выявляется. Однако после 2 -4 летнего возраста появляются симптомы андрогенизации: Оволосение в аксиллярных областях и на лобке Маскулинизация фигуры за счет быстрого развития скелетной мускулатуры

Вирильная форма Мутация голоса, появляются юношеские угри Происходит увеличение полового члена Ускоряется дифференцировка скелета (костный возраст опережает паспотрный на 2 и более лет) Зоны роста закрываются преждевременно (к 9 -10 годам), что обусловливает конечную низкорослость

Диагностические критерии АГС АД – норма Глюкоза – норма РН – норма К, Na, СL – норма КОРТИЗОЛ – снижен АКТГ – повышен 17 -он – ПРОГЕСТЕРОН – повышен Тестостерон –повышен 17 - КС в суточной моче повышены (метаболиты андрогенов) Ренин – умеренно повышен
: в пре- и пубертатном периоде. У девочек появляется вирильный синдром")
НЕКЛАССИЧЕСКАЯ ФОРМА (ПУБЕРТАТНОГО ПЕРИОДА): в пре- и пубертатном периоде. У девочек появляется вирильный синдром (преждевременное адренархе, позднее менархе, умеренный гирсутизм, ускорение роста). Может формироваться поликистоз яичников. У мальчиков клинические проявления отсутствуют. В отдельных случаях возникает гинекомастия.
: Физическое развитие – опережает Костный возраст - опережает Кортизол –")
НЕКЛАССИЧЕСКАЯ ФОРМА (ПУБЕРТАТНОГО ПЕРИОДА): Физическое развитие – опережает Костный возраст - опережает Кортизол – норма АКТГ – повышен 17 -КС повышены Тестостерон – повышен 17 -он-прогестерон – норма или умеренно повышен Электролиты , АД, РН, ренин , глюкоза - норма

СОЛЬТЕРЯЮЩАЯ ФОРМА: Развивается в случае значительного дефицита 21 -гидроксилазы. При этом, помимо гиперандрогенемии, нарушается адекватный синтез кортизола и альдостерона. У НВ девочек – ву вирилизация. У НВ мальчиков – увеличение наружных половых органов, их пигментация.

СОЛЬТЕРЯЮЩАЯ ФОРМА: На этом фоне возникают клинические симптомы острой надпочечниковой недостаточности, которые появляются с первых дней жизни (с 7 -30 дня): Срыгивания, рвота «фонтаном» не связана с приемом пищи (ДД с пилоростенозом) Диарея Потеря массы тела Симптомы эксикоза (кожа сухая, сероватого цвета, с мраморным рисунком, сухость слизистых оболочек)

СОЛЬТЕРЯЮЩАЯ ФОРМА: Снижение тургора тканей и эластичности кожи Западение большого родничка Нt – повышен (гемоконцентрация) Гемодинамические нарушения (снижение АД, тахикардия, глухость сердечных тонов, мраморность кожи, цианоз) Адинамия, слабость, выраженная мышечная гипотония Гиперпигментация (области складок, ареолов и т. д. )

СОЛЬТЕРЯЮЩАЯ ФОРМА: Электролитные нарушения: - гиперкалиемия, - гипонатриемия, - гипохлоремия Гипогликемия Метаболический ацидоз Кортизол – снижен АКТГ – повышен 17 -он-прогестерон – повышен Тестостерон – повышен 17 - КС – повышены (метаболиты андрогенов) Альдостерон – понижен Ренин – повышен

ГИПЕРТОНИЧЕСКАЯ ФОРМА: Генетический локус фермента 11 гидроксилазы CYP 11 B 1 находится на 8 q. Девочки: симптомы грубой вирилизации (ложный женский гермафродитизм) Мальчики: ППР и ускоренное физическое развитие (маскулинизация телосложения)

ГИПЕРТОНИЧЕСКАЯ ФОРМА: Раннее и стойкое повышение АД т. к. повышены уровни 11 -дезокси-кортикостерона и 11 дезокси-кортизола Кортизол – снижен АКТГ – повышен 17 -он-прогестерон – повышен
 Ренин –")
ГИПЕРТОНИЧЕСКАЯ ФОРМА: Тестостерон – норма 17 - КС – повышены (метаболиты андрогенов) Ренин – снижен Гипокалиемия Гипернатриемия ХЛОР – повышен или норма Глюкоза норма РН -норма
 Глюкокортикоиды: гидрокортизон 15 -25 мг/м 2 (подросткам 30 -40 мг/м")
ЛЕЧЕНИЕ АГС (вирильная форма) Глюкокортикоиды: гидрокортизон 15 -25 мг/м 2 (подросткам 30 -40 мг/м 2) Или преднизолон 3 -5 мг/м 2 , (подросткам – 6 мг/м 2 ) Разделить на 2 -3 приема: 6 -8 00 - 50% 12 -14 00 - 25 % 17 00 - 25 %
 Хирургическое лечение: Начинают с 3 -ей степени вирилизации: Первый этап –")
ЛЕЧЕНИЕ АГС(вирильная форма) Хирургическое лечение: Начинают с 3 -ей степени вирилизации: Первый этап – клитерэктомия и формирование малых половых губ (до 2 -х летнего возраста); Второй этап – пластика влагалища после 10 -12 -летнего возраста.
: ДЕКСАМЕТАЗОН – 18 – 12 табл. 1 раз на")
ЛЕЧЕНИЕ НЕКЛАССИЧЕСКОЙ ФОРМЫ (ПУБЕРТАТНОГО ПЕРИОДА): ДЕКСАМЕТАЗОН – 18 – 12 табл. 1 раз на ночь При отсутствии эффекта – ципротерона ацетат (антиандрогены)
: гидрокортизон 10 -20 мг/кг в/в струйно (или")
ЛЕЧЕНИЕ СОЛЬТЕРЯЮЩЕЙ ФОРМЫ: 1. Заместительная терапия (глюкокортикоиды): гидрокортизон 10 -20 мг/кг в/в струйно (или преднизолон 2 -4 мг/кг) - далее гидрокортизон по 2 -4 мг/кг в/в каждые 4 часа (т. е. 6 раз в сутки) Минералокортикоиды перорально (после прекращения рвоты): - флудрокортизон (кортинеф) - 0, 1 - 0, 2 мг/сутки

ЛЕЧЕНИЕ СОЛЬТЕРЯЮЩЕЙ ФОРМЫ: 3. Регидратация: - 5% раствор глюкозы и 0, 9 % раствор Na. Cl в соотношении 1: 1 в объеме 50 мл/кг за первые 1 -2 часа терапии, далее по 25 мл/кг - за последующие 3 -4 часа инфузию продолжать по 20 -25 мл/кг в зависимости от состояния.

ЛЕЧЕНИЕ СОЛЬТЕРЯЮЩЕЙ ФОРМЫ: При выраженном снижении артериального давления вводят допамин со скоростью 8 -10 мкг/кг/мин. (предварительно развести с 200 мл 0, 9 % раствора Na. Cl) Можно использовать 0, 2 % раствор норадреналина 40 -50 капель/мин. Контроль артериального давления осуществляют каждые 5 -10 минут.

ЛЕЧЕНИЕ ГИПЕРТОНИЧЕСКОЙ ФОРМЫ: Глюкокортикоиды: преднизолон 3 -5 мг/м 2 , (подросткам – 6 мг/м 2 ) Минералокортикоиды перорально (после прекращения рвоты): - флудрокортизон (кортинеф) - 0, 025 - 0, 05 мг/ м 2 1 раз в сутки Гипотензивные препараты – подбор индивидуально.
")
МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ (МБ)
 генетические и структурно-биохимические дефекты митохондрий, которые сопровождаются")
Общая характеристика митохондриальной патологии Митохондриальные болезни (цитопатии) генетические и структурно-биохимические дефекты митохондрий, которые сопровождаются нарушением тканевого дыхания.
 Поражение энергозависимых тканей: - мозг мышцы миокард")
Патогенез МБ: Дефект энергетического метаболизма (окислительного фосфорилирования) Поражение энергозависимых тканей: - мозг мышцы миокард - печень почки панкреас - орган зрения

Митохондриальный геном: Клетка содержит: сотни – тысячи митохондрий В митохондриях - митохондриальная ДНК (мт. ДНК) - это кольцевая молекула ДНК, которая включает 16. 569 нуклеотидов мт ДНК содержит 37 генов, которые кодируют: - 2 вида рибосомальной РНК - 22 вида транспортной РНК - 13 белков дыхательной цепи митохондрий - (остальные белки дыхательной цепи кодируются ядерной ДНК)

Митохондрии передаются с цитоплазмой ооцитов. Спермии не имеют митохондрий, т. к. цитоплазма элиминируется при созревании мужских половых клеток При клеточном делении: митохондрии в случайном порядке переходят в дочернии клетки. При мутации процентное содержание нормальной и мутантной мт. ДНК в разных тканях варьирует от 0% до 100%
 наследование: Болеют все дети (сестры и братья)")
Типы наследования МБ: 1. Материнское (митохондриальное, цитоплазматическое) наследование: Болеют все дети (сестры и братья) , рожденные от больной матери Синдром MELAS Синдром MERRF Синдром NARP Атрофия зрительных нервов Лебера
 наследование (мутация в ядерной ДНК): Аутосомно-доминантный")
Типы наследования МБ: 2. Менделевское (ад, ар, Х-сцепленное) наследование (мутация в ядерной ДНК): Аутосомно-доминантный тип: (50 % больных от больной матери или от больного отца) Аутосомно-рецессивный тип: 25 % больных (рожденных от гетерозиготных носителей)

Х-сцепленный рецессивный: 50% сыновей – больные 50% дочерей – носители Болезнь Фридрейха Болезнь Лея Синдром Альперса

Спорадические случаи: Отсутствуют повторные случаи болезни в семье - Синдром Кернса-Сейра - Прогрессирующая наружная офтальмоплегия

Тяжесть и характер клиники коррелируют : С тяжестью мутации мт. ДНК С процентным содержанием мутантной мт. ДНК в конкретных тканях С порогом чувствительности тканей к дефектам тканевого дыхания (ГМ, миокард – наиболее зависимы от аэробного дыхания; Кожа, кости, хрящевая тк. , лимф. с-ма – низкая чувствительность)

Диагностика МБ: 1. Полисистемность поражения 2. ЛАКТАТ-АЦИДОЗ: - повышение лактата и пирувата в крови и СМЖ 3. Феномен «рваных красных волокон» в миофибриллах ( это пролиферация митохондрий)
 5. Электронная микроскопия: -")
Диагностика МБ: 4. Дефицит цитохром-С-оксидазы в мышечных волокнах (гистохимическое исс) 5. Электронная микроскопия: - аномалия формы, размеров, - наличие включений в митохондриях
 Точечные мутации в мт. ДНК в нуклеотидах 3243 и 3271")
Синдром MELAS (митохондриальная энцефаломиопатия) Точечные мутации в мт. ДНК в нуклеотидах 3243 и 3271 Чем больше накоплено мутаций , тем тяжелее заболевание пг: острая недостаточность энергетических субстратов в клетках И высокая чувствтельность головного мозга к токсическим влияниям
")
Клинические симптомы Заболевание проявляется в 6 -10 лет (в отдельных случаях: 2 -40 лет) Рецидивирующие головные боли Рвота Анорексия Непереносимость длительной физической нагрузки: - резкая мышечная слабость - миалгии

Инсультоподобные эпизоды: Головные боли Головокружения Обмороки Неврологическая симптоматика: Гемипарезы, гемианопсии Коматозные состояния
 Постепенно развивается деменция Возможны :")
Судороги: Фокальные пароксизмы Генерализованные клонико-тонические (Судороги резистентны к антиконвульсантам) Постепенно развивается деменция Возможны : сахарный диабет, гипопаратиреоз АВ – блокады, синдром WPW Атрофия зрительных нервов Тугоухость Мозжечковый синдром

Диагностические критерии Материнский тип наследования Возраст манифестации до 40 лет Мигренозные пароксизмы с тошнотой и рвотой Инсультоподобные эпизоды Судороги

Диагностические критерии В крови: лактат-ацидоз В моче: ацидурия КТ головного мозга: кальцификация базальных ганглиев Биопсия скелетных мышц: «рваные красные волокна» Прогрессирующее течение (глубокая инвалидизация) Лечение: метаболическая терапия

Синдром MERRF В основе – точечные мутации в гене лизиновой т. РНК в позиции 8344 и 8356 мт. ДНК Манифестация в различные возрастные периоды : от 6 до 65 лет - Быстрая утомляемость при физической нагрузке - Боли в икроножных мышцах - Снижение памяти, внимания

Типичный симптомокомплекс: 1. Прогрессирующий миоклонус : Неожиданные быстрые короткие мышечные сокращения Деменция Атаксия 2. Генерализованные тонико-клонические судороги Сенсорная глухота Атрофия зрительного нерва

Критерии диагностики Материнский тип наследования Клинические проявления Лактат-ацидоз Умеренное повышение белка в ликворе Недостаточность 1, 3, 4 комплексов дыхательной цепочки

Критерии диагностики КТ: атрофия мозга, лейкоэнцефалопатия, кальцификация базальных ганглиев Биопсия скелетных мышц: симптом «рваных красных волокон» ЭЭГ: генерализованные «спайк-волны» ЭМГ: первично-мышечный тип поражения

Лечение синдрома MERRF Коррекция нарушений энергетического обмена Коррекция лактат-ацидоза Рибофлавин Никотинамид Цитохром С Коэнзим Q 10

Лечение синдрома MERRF L-карнитин Витамин С Антиконвульсанты –вольпроаты по 30 мгсутки В случае неэффективности- клоназепам
Генные болезни Дивинская.ppt