d0b342df8eecbc01579013a274271fcb.ppt
- Количество слайдов: 24



Faculdade de Medicina de Ribeirão Preto Disciplina: Genética Humana Docente: Profa. Dra. Aparecida M. Fontes Uma Aliviante Lufada de Ar Fresco Compreensão Fundamental da biologia básica abre espaço para novos tratamentos em fibrose cística. Ana Paula Fonte Leticia Freitas Borges Stéfani Abreu de Morais Ribeirão Preto 2014.

Publicado pela SCIENTIFIC AMERICAN BRASIL. Edição: ESPECIAL SAÚDE 1. Em Agosto/ Setembro de 2013. • Steven M. Rowe é professor- assistente de medicina, pneumologia, pediátrica físiologia e biofisica da University of Alabama, em Birminfham; • J. P. Clancy é professor de medicina pulmonar pediátrica do Cincinnati Children’s Hospital Center e da University of Cincinnati; • Eric J. Sorscher é professor de medicina e fisiologia e biofisica da University of Alabama, em Birmingham.

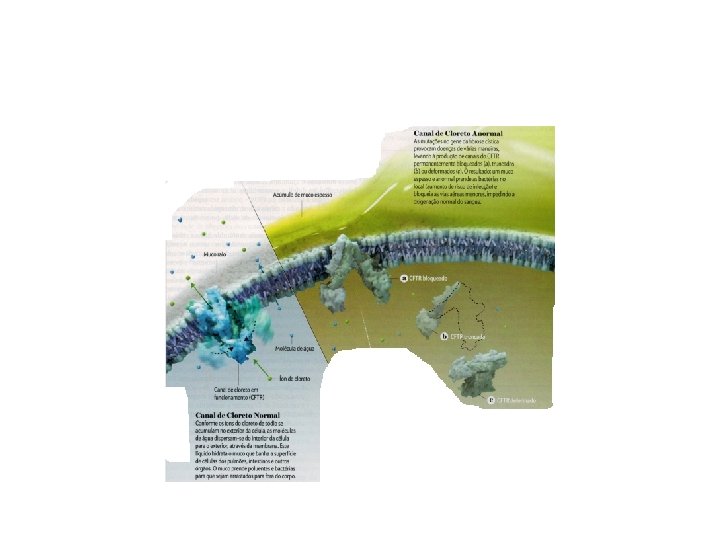
O que é Fibrose Cística? • A Fibrose Cística ou Mucoviscidose é uma doença hereditária autossômica recessiva que afeta a produção e fluxo do muco dentro dos pulmões e no sistema digestivo; as secreções impedem a passagem das enzimas digestivas deixando as áreas obstruídas com o muco pegajoso. • É Falha na capacidade no transporte de cloreto de sódio. • Comum na população caucasiana, aparece aproximadamente 1: 2. 500 nascimentos.

Teste Padrão da Herança para a Fibrose Cística

§ Atualmente com o avanço nos estudos científicos, os pacientes chegam quase aos 40 anos, porém antes do uso de antibióticos para tratar infecções pulmonar as crianças ainda morriam na infância.

§ A fibrose cística normalmente é diagnosticada na infância, pelos programas de triagem neonatal ou pelo teste do suor.

Causas Gene CFTR descoberto em 1989;

A mutação do gene resulta em mal-funcionamento do canal de CFTR (Regulador de condutância transmembranar de fibrose cística) que transporta íons cloreto.
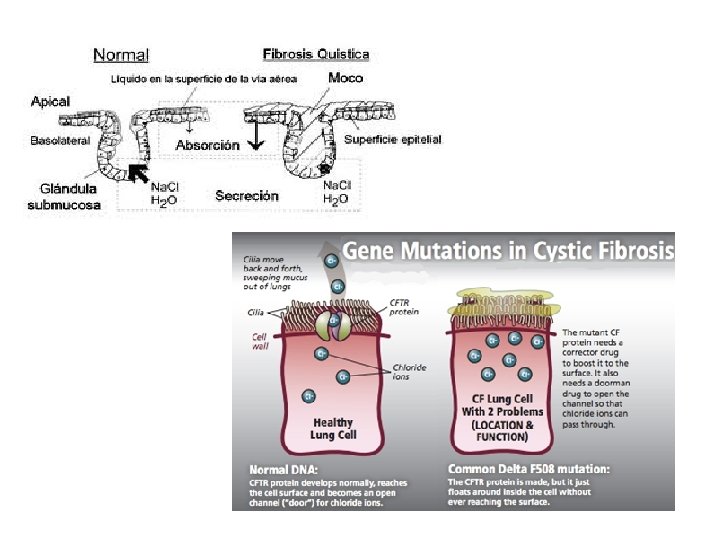

• Causa genética mais comum mutação F 508 del

• A mutação genética mais comum no canal de cloreto de sódio é totalmente ausente na superfície da célula que ocorre na exclusão de apenas um dos canais de 1. 500 blocos de construção de aminoácidos, no caso a FENILALANINA (F 508), conhecida como F 508 del.

• Mutações que resultam em canais encurtados de CFTR representam 10% dos casos de FC; • Defeito genético W 1282 X é responsável por cerca de 40% das FC em Israel; • Proteína acaba truncada porque o gene recebe instruções equivocadas e ordenadas do mecanismo de síntese de proteínas da posição 1282 da cadeia a qual o aminoácido triptofano normalmente está presente;

• O conjunto de mutações que desativa a capacidade de abertura do canal responde a 5% dos casos de FC do mundo; • Compostos projetados para manter a porta do CFTR mutante que pode ajudar os pacientes.
 localizada no")
• A imagem a esquerda mostra o CFTR não mutado (verde) localizada no interior da célula, ou seja, no citosol, e a membrana citoplasmática, que se situa na periferia da célula. O núcleo de cada célula é simbolizado em azul. • A imagem da direita mostra a proteína mutante F 508 del agrupada em torno do núcleo, em vez da membrana. Esta diferença na localização é o centro do problema.

Tipos de Tratamento para FC • Manutenção do estado nutricional; • Prescrição de suplementos energéticos; • Dietas hiperlípidicas e hiperprotéicas, bem como a suplementação de minerais e vitaminas lipossolúveis. • A remoção das secreções das vias aéreas com fisioterapia e medicamentos que ajudam a diluir o muco; • Uso de antibióticos para prevenção e tratamento de infecções; • Assistência Multidisciplinar; • Suporte de apoio a família;

Tipos de Tratamento para FC

Candidatos a Fármacos Promissores Drogas foram criadas a partir da biologia básica da Fibrose Cística. • Vertex Pharmaceutical, VX-770 – Mutação G 551 D – Abre o canal em CFTR bloqueadas; • Vertex Pharmaceutical, VX-809 – Mutação F 508 del - Correção e encaminhamento da proteína para membrana; • PTC Therapeutics, Ataluren - Mutações menos comuns – instruções equivocadas de “parada”; • Combinação de corretores com potenciadores; • Diferentes drogas deverão ser criadas.

Contribuição da Fonoaudiologia no tratamento de FC • Atuação fonoaudiológica em exercícios respiratórios (Lemos & Oliveira, 2006). • Perda auditiva significante em pacientes com FC (Martins LM et al. , 2010).

Considerações Finais • https: //www. youtube. com/watch? v=0 NMQ-i. Ic 4 co

Perspectiva nos estudos sobre Fibrose Cística • Causa subjacente da doença e não os sintomas. • Terapia Gênica - Por que os vírus não executaram bem o trabalho? • Interferir no controle de qualidade da célula não acarretaria em alguma outra disfunção? • Aconselhamento genético. • Teste do pézinho. •

Referências Bibliográficas • ROWE. STEVEN. M; CLANCY J. P. ; SORSCHER ERIC J. Uma aliviante lufada de ar fresco. Scientific American Brasil; Edição especial saúde 1, 54 ; 26 -31, 2013. • ROSA. FERNANDA. R; FERNANDA. DIAS. G; LUCIANA. NOBRE. N; HARRIMAN. MORAIS. A. Fibrose cística uma abordagem clínica e nutricional. Scielo Brasil; vol. 21, 6; 2008. • <http: //www. hospitaldopulmao. com. br/site/hospital/exames-orientacoes> Acesso em 21 de Outubro de 2014. • <http: //drauziovarella. com. br/letras/f/fibrose-cistica/> Acesso em 15 de Outubro de 2014. • <http: //aprovaja. blogspot. com. br/2012/05/teste-do-pezinho-para-todos-os-bebes. html> Acesso em 21 de Outubro de 2014. • <http: //www. news-medical. net/health/Cystic-Fibrosis. Causes(Portuguese). aspx>Acesso em 12 de Outubro de 2014.

• <http: //www. hermespardini. com. br/atual_manual/pdf_genetica_novos_exames/ Fibrose_Cistica_-_Estudo_Genetico. pdf > Acesso em 31 de Outubro de 2014. • CABELLO, GMK. Avanços da genética na fibrose cística. Revista Hospital Universitário Pedro Ernesto, 10(4), 2011. • REIS, FJC; DAMACENO, N. Fibrose Cística. Jornal de Pediatria, 74 (supl. 1): S 76 – S 94, 1998. • <http: //www. ncbi. nlm. nih. gov/gene/1080> Acesso em 31 de Outubro de 2014. • CARTER, JM; JOHNSON, BT; PATEL, A; PALACIOS, E; RODRIGUEZ, KH. Lund-mackay staging system in cystic fibrosis: A prognostic factor for revision surgery? The Ochsner Journal, 14(02): 184 - 187, 2014.

Obrigada !
d0b342df8eecbc01579013a274271fcb.ppt