5c11baec5489399d1f32b4fec487ec08.ppt
- Количество слайдов: 73



Expanded Newborn Screening: Public Health Policy and Clinical Impact Nutrition 526 October 18, 2010 Beth Ogata, MS, RD, CSP, bogata@uw. edu Cristine M Trahms, MS, RD, FADA

Newborn Screening A state mandated public health program that begins with a “heel poke” for every baby before hospital discharge First screen must be taken 24 -48 hours of life regardless of feeding status or weight Blood Sample on Guthrie Filter Paper Card

Who is screened? Washington State law requires that every newborn be tested prior to discharge from the hospital or within five days of age Second screen strongly recommended between 7 and 14 days of age) Third screen recommended for sick and premature infants

Why do newborn screening? Screen a presumably healthy newborn population Detect disease before symptoms present clinically Goal: Prevent or reduce morbidity and mortality

Criteria for Newborn Screening Important condition Acceptable treatment available Facilities for diagnosis and treatment Difficult to recognize early Suitable screening test Natural history known Cost-effective to diagnose and treat Wilson & Jungner, 1968
 High Impact and High Throughput One disease, one test is")
Tandem Mass Spectrometry (MS/MS) High Impact and High Throughput One disease, one test is not cost-effective Many diseases, one test is cost-effective MS/MS allows for rapid, simultaneous analysis and detection of many disorders of amino acid, organic acid, and fatty acid metabolism
")
Tandem Mass Spectrometer (MS/MS)
 Stable isotope internal standards added")
MS/MS Methodology Blood spots punched (3/16 th inch disc) Stable isotope internal standards added (deuterated) Butyl esters derivatives made Automatic injection into MS/MS via 96 well plates Sample set up determines which masses and therefore which compounds are detected 2 minute analysis time Automated data processing for results

MS/MS Methodology – continued Compounds analyzed are amino acids and acylcarnitines Amino acids – to identify PKU, MSUD, homocystinuria n Acylcarnitine – carnitine (vehicle) + fatty acid for identification of organic acidurias and fatty acid oxidation disorders n

MS/MS Plasma Acylcarnitines 100% C 8 Intensity * C 2 MCAD * * C 6 * C 10: 1 * * C 16 100% Control Intensity * C 2 * * * internal standards

MS/MS Plasma Amino Acids

What is the scope of newborn screening? Screen ~80, 000 newborns Receive ~160, 000 specimens Track ~3000 infants with abnormal results Prevent ~140 babies from death or disability For example: In WA State

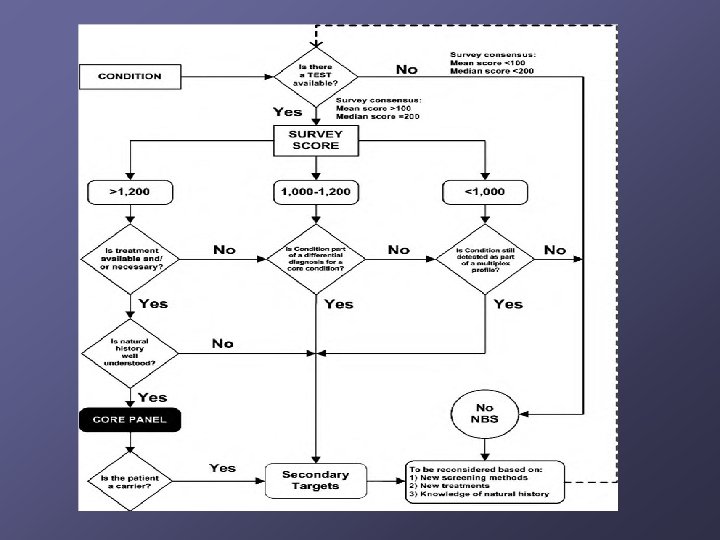
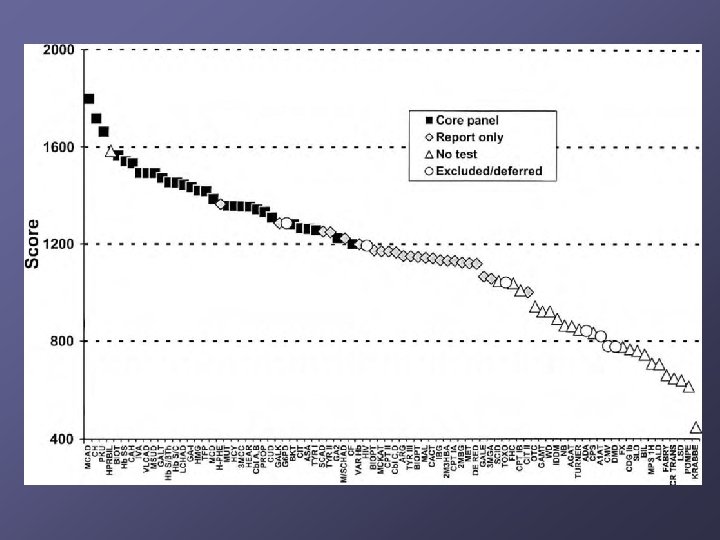
Which disorders should be identified? NBS mandates are under state control n Some states screened for 3 diseases, others 40+ 2002 Maternal and Child Health Bureau commissioned ACMG n n n Analyze literature Develop consensus on which disorders Recommend a core panel to create uniform NBS across all states

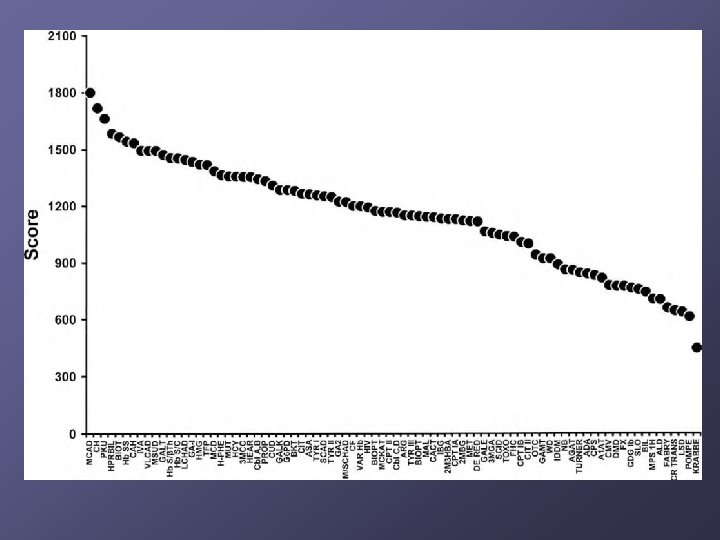
 Early PKU screening led to cases of overrestriction and/or implementation")
Historical Harm (? ) Early PKU screening led to cases of overrestriction and/or implementation of diet prior to confirmation of diagnosis n n Today, diagnosis is quite rapid 40 years ago, it took much longer so more potential for harm However, no published evidence of widespread physical/medical harm BUT the cases do underscore need for expertise and resources for management

Amino Acid Disorders AA that are not used to make proteins are recycled by their specific metabolic pathways. n Enzymatic deficiencies in these pathways lead to various clinical phenotypes. Diagnosed by plasma amino acids, urine amino acids, and/or urine organic acids (takes 2 -5 days) PKU: severe, permanent ID MSUD: ID, hallucinations, ataxia HCY: connective tissue damage (joints, heart), ID, psychiatric disturbances CIT: risk of hyperammonemia ID, coma, death ASA: brittle hair, liver disease ID TYR I: acute or chronic liver disease, liver cancer, neurologic pain crises

Organic Acid Disorders Organic acids are breakdown products of protein and fatty acid metabolism. Defects in their breakdown lead to (generally): n n Vomiting, metabolic acidosis, elevated ammonia in crises ID, motor delay, ataxia, cardiac/renal/pancreatic problems Diagnosed by urine organic acids and/or plasma acylcarnitines IVA: Isovaleric acidemia GA I: Glutaric acidemia type I HMG: 3 -OH 3 -CH 3 glutaric aciduria MCD: Multiple carboxylase deficiency MUT: Methylmalonic acidemia (mutase deficiency) 3 MCC: 3 -Methylcrotonyl-Co. A carboxylase deficiency Cbl A, B: Methylmalonic acidemia PROP: Propionic acidemia BKT: Beta-ketothiolase deficiency

Fatty Acid Disorders Fatty acid disorders lead to impaired energy production n Hypoglycemia, cardiomyopathy, muscle weakness can be seen Diagnosed by plasma acylcarnitines, and urine organic acids can be helpful MCAD: Medium-chain acyl-Co. A dehydrogenase deficiency VLCAD: Very long-chain acyl-Co. A dehydrogenase deficiency LCHAD: Long-chain L-3 OH acyl-Co. A dehydrogenase deficiency TFP: Trifunctional protein deficiency CUD: Carnitine uptake defect

Who is identified? 1. Patients who need active management n n n Symptomatic at diagnosis Strong evidence of pathology if untreated Examples: PKU, classic galactosemia, MSUD, PROP, etc.

Who is identified? 2. Patients with disorders known to pose risk but reduced penetrance n n i. e. , probably not everyone needs to be treated HPHE, MCAD Both are/have mild ends of the spectrum that have only been identified through NBS MCAD mutation c. 199 C>T Never seen in patients picked up clinically

Who is identified? 3. Patients who may not need any management n Disorders considered extremely rare but seen in large numbers via NBS programs Reported cases have significant morbidity NBS pickups are mostly mild 3 MCC, SCAD n Biochemical phenotype
 Proceeding with caution Not screening Core diseases")
Proceeding with Caution (Reasons to be Thoughtful) Proceeding with caution Not screening Core diseases vs. secondary targets / unintended targets n n n What is reported vs. withheld? Will we pick up untreatable conditions? What is the impact of false positives on families? No long-term outcome data – consider research paradigm Consider infrastructure needed for follow-up

What are we screening for? 9 OA 5 FAO 3 Hb Pathies 6 AA 6 Others CORE PANEL IVA GA I HMG MCD MUT 3 MCC Cbl A, B PROP BKT MCAD VLCAD LCHAD TFP CUD PKU MSUD HCY CIT ASA TYR I Hb SS Hb S/ßTh Hb S/C CH BIOT CAH GALT HEAR CF

How many infants does NBS identify? 2006 2007 Infants Diagnosed 2 1 Biotinidase deficiency 5 5 Congenital adrenal hypoplasia (CAH) 45 45 Congenital hypothyroidism (CH) 12 14 Cystic fibrosis 6 0 Galactosemia 1 0 Homocystinuria 0 0 Maple syrup urine disease 3 6 Medium chain acyl co. A dehydrogenase (MCAD) def. 7 7 Phenylketonuria (PKU) 13 23 Sickle cell and other HG 95 112 TOTAL

Emma 13 months old, healthy Normal pregnancy and delivery Normal eating pattern, no allergies or intolerances Feb 2008: n n Vomited 4 -5 times throughout the weekend No fever Sleeping for extended periods – parents concerned, but previous fever had same pattern Parents gave Pedialyte

Emma 4½ yo brother, parents sick on Sunday/Monday; same symptoms Monday night 9: 30 checked on Emma n Raspy breathing – thought respiratory problem but not worried Tuesday morning 11 am she was found motionless in her crib and pronounced dead at the scene

Emma Autopsy revealed fatty changes to liver Coroner requested newborn screening blood spot be sent for acylcarnitine profile Diagnostic for very long chain acyl-co A dehydrogenase deficiency (VLCAD)

VLCAD Disorder of long chain fatty acid breakdown C 14, C 14: 1 C 16, C 18 Normal beta oxidation occurs in mitochondria

Fatty Acid Oxidation http: //www. genomeknowledge. org/figures/saturatedbetao. jpg

VLCAD Presentations Hypertrophic cardiomyopathy, with hypoglycemia and skeletal myopathy, lethargy, failure to thrive n Usually present birth to 5 months Hypoglycemia, hepatomegaly, muscle weakness without cardiac manifestations n Late infancy – older childhood Muscle weakness/pain, rhabdomyolysis with exercise or illness. No hypoglycemia or cardiac n Teens to adulthood

VLCAD Treatment Diet low in long-chain fats (Portagen, Monogen = 87%, 90% of fats as MCT) Additional medium chain fats (MCT oil, walnut oil) Carnitine 100 mg/kg/day Avoidance of fasting Treating illness with IV glucose support
 DNA")
VLCAD Diagnosis Newborn screening Plasma acylcarnitine profile Urine organic acids (should be normal) DNA sequencing
")
Emma’s Family referred to genetics by coroner Parents requested testing for older brother (Zach) Acylcarnitine ordered DNA sequencing of ACADVL gene ordered
 C 14: 1 C 14 C 16 - nl")
Acylcarnitine – Zach (5 yo) C 14: 1 C 14 C 16 - nl C 16: 1 - nl

Zach Testing Reported: mild elevation of C 14 and C 14: 1 with low free carnitine. VLCAD cannot be ruled out Recommend supplementing with carnitine and retest in 1 week DNA testing results back before AC repeat: Zach’s DNA testing reveals he is affected Family seen in clinic, started on treatment

Zach – Clinical Picture 5 yo Healthy No symptoms of muscle weakness n CPK = 315 U/L (35 -230) No hepatomegaly n n n AST= 49 (5 -41) ALT= 23 Bilirubin conj, unconj = normal (0. 0, 0. 4) No evidence of cardiac involvement Has had several viral illnesses in his lifetime without difficulty Once on carnitine, AC profile was classic for VLCAD

Components of Newborn Screening Sampling n hospital partnerships Screening n State Lab Reporting n to health care provider Referral n to specialty care provider Short term follow-up n diagnosis Long term follow-up n ongoing treatment & monitoring

Washington State Newborn Screening Birth Day 1 First Screen + NL ++ Primary Doctor 2 nd Sample NL DX Primary Care Doctor/ Biochem Clinic ASAP TX + Primary Care Doctor Biochem Clinic Long term Follow up DX • Timely/urgent • Systematic process TX Long term Follow up

Effective NBS requires a close working relationship between hospitals, newborn screening program, and follow-up program Informed Consent

Supporting understanding for families

Nutrition Involvement in NBS Policy Diagnostic/coordination Clinical Community

Example: infant with galactosemia Symptoms in newborn, if untreated n n n n Vomiting, diarrhea Hyperbilirubinemia, hepatic dysfunction, hepatomegaly Renal tubular dysfunction Cataracts Encephalopathy E. coli septicemia result Death within 6 weeks, if untreated Also n n n Duarte variant galactokinase deficiency uridine diphosphategalactose-4 -epimerase deficiency Galactose-1 -phosphate uridyl transferase (GALT) deficiency

Example: infant with galactosemia Treatment: eliminate all galactose from diet Primary source is milk (lactose= galactose + glucose) Secondary sources are legumes Minor? sources are fruits and vegetables Food labels n milk, casein, milk solids, lactose, whey, hydrolyzed protein, lactalbumin, lactostearin, caseinate Medications (lactose is often an inactive ingredient) Dietary supplements Artificial sweeteners Monitoring: galactose-1 -phosphate levels <3 -4 mg/dl

Example: Infant with galactosemia POLICY CLINICAL MANAGEMENT RD participated on State Advisory Board to select disorders, including galactosemia RD provides nutrition care as member of the Biochemical Genetics Team: • Initiation of formula • Guidelines for monitoring intake • Plans for follow-up DIAGNOSIS & COOORDINATION “Presumptive positive” RD in contact with family and local providers to discuss appropriate feeding practices and arrange clinic appointment RD as case manager COMMUNITY RD at local health department provides ongoing education to family, local care providers
 RD participated in Advisory Board meetings,")
Nutrition and NBS: Policy Screening process (disorders, procedures) RD participated in Advisory Board meetings, providing input about nutrition-related treatment Services and reimbursement Nutrition consultant to state CSHCN Program RD provides input about relevant state Medicaid policies Training and education RD provides information about management of metabolic disorders to local WIC agencies

Nutrition and NBS: Clinical Management – PKU Phenylketonuria n n n Phenylalanine hydroxylase Dihydropteridine reductase Biopterin synthetase Establish diagnosis n Presumptive positive NBS results > 3 mg/d. L, >24 hrs of age n Differential diagnosis serum phe, nl tyr r/o DHPR, biopterin defects

Current Treatment Guidelines With effective NBS, children are identified by 7 days of age Initiate treatment immediately Maintain phe levels 1 -6 mg/dl (60 -360 umol/L) Lifelong treatment

Outcome Expectations With NBS and blood phenylalanine levels consistently in the treatment range n Normal IQ and physical growth are expected With delayed diagnosis or consistently elevated blood levels n IQ is diminished and physical growth is compromised

Clinical Management: PKU Goals of Nutrition Therapy Normal growth rate Normal physical development Normal cognitive development Normal nutritional status

Clinical Management: PKU Correct substrate imbalance n Restrict phenylalanine intake to normalize plasma concentration Supply product of reaction n Supplement tyrosine to maintain normal plasma tyrosine levels Phenylalanine ----------//------------ Tyrosine (substrate) phenylalanine hydroxylase (product)

Phe Levels from NBS to Tx Diagnostic levels Blood levels every 2 days because of rapid growth Equilibrium achieved by 14 days of age

Adjustments necessary to maintain “safe” blood phe levels Usual intake of phe n Newborn on formula 20 oz x 22 mg phe/oz = 440 mg phe n 1 yo child on “regular” diet 30 g protein = 1500 mg phe (DRI = 13. 5 g) n 7 yo child on “regular” diet 50 g protein = 2500 mg phe (DRI = 19 g) Phenylalanine requirement n 250 mg/d

Management Tools Specialized formula provides n n 80 -90% energy intake 89 -90% protein intake tyrosine supplements no phenylalanine Phenylalanine to meet requirement from infant formula or foods

Food Choices for PKU

Effect of a single amino acid deficiency on growth

Effective Blood Level Management in Childhood Blood levels once per month, or more frequently if needed for good management

PKU Management Guidelines Selfmanagement Skills

Goal of Lifetime Management of PKU To maintain metabolic balance while providing adequate nutrients and energy for normal physical and intellectual growth

Maternal PKU Concerns/Outcomes Women with PKU are at high risk for delivering a damaged infant n Placenta concentrates phe 2 -4 x Microcephaly Cardiac problems Infant IQ directly related to maternal blood phe level Outcome improved with maternal blood phe <2 mg/dl prior to conception and during pregnancy

Nutrition and NBS: Community PHN and interpreter make monthly visits to family of young child with MSUD. Through pre-arranged phone calls, we can discuss formula composition and preparation, and solid foods. This helps provide information between regular clinic visits.

Nutrition and NBS: Community A woman with PKU is enrolled in the First Steps program (WA State MSS). The RD with PKU Clinic provides consultation to the First Steps RD, about management of amino acid levels.

Metabolic Team Child Age-appropriate self-management skills Parents Monitoring health status, teaching, advocacy Nutritionist Nutrition therapy, feeding skills Geneticist Medical monitoring Social Worker Family support, counseling Laboratory monitoring Medical Home Well child care, family support Psychologist Developmental monitoring, counseling PHN, others Family support in community School Educational programs, treatment monitoring Community Support of family and friends Therapists (OT, PT, SLP, etc. ) Developmental monitoring, intervention

NBS and the Community: Challenges Understand the implications of the results of newborn screening tests Develop a communication system between the community providers and the metabolic team for support of treatment Interact with PCPs and families as needed, to support appropriate MNT

NBS and the Community: What you need to know Which disorders are identified by NBS in your state? Where do you find this information? What is the difference between screening and diagnostic results? What is the system for follow-up of presumptive positive NBS results? How do you make referrals to regional genetics clinics and specialty care clinics?

Scenes from the Annals of Reporting and Acting on NBS Results A primary care physician telephones are reports there is a new baby with PKU and asks that you please start the infant on formula ASAP. What additional information do you need? What would you do?

Scenes from the Annals of Reporting and Acting on NBS Results You are on-call for the weekend for your local hospital and you receive an order from the newborn nursery on an infant with presumptive galactosemia and a request for the initiation of treatment. What additional information do you need? What would you do?

Summary NBS is the first part of a process of care that requires strong partnerships for optimal outcomes NBS outcomes are only as good as the follow-up provided Families should have access to the best treatment and care for their child

Summary Specific diagnosis must be confirmed n in coordination with the state Newborn Screening Program Careful monitoring of medical and nutritional status must be on-going n by the metabolic team Nutritional intervention n n must be specific to the disorder specific to the child

Additional Information Washington State Newborn Screening http: //www. doh. wa. gov/ehsph/phl/newborn/default. htm National Newborn Screening and Genetics Resource Center http: //genes-r-us. uthscsa. edu Star G-Screening, Technology, and Research in Genetics http: //newbornscreening. info Building Block for Life (PNPG) n n Expanded NBS – 27(1) Genetics and Expanded NBS – 30(3) Nutrition Focus n n Overview nutr assessment of children with metabolic disorders – 24(5) Genetics – 22(6) Journal of Developmental and Behavioral Pediatrics n Levy PA. An overview of newborn screening. 2010; 31(7): 622.

Why do we do newborn screening? So Super Girl can be whoever she wants to be….
5c11baec5489399d1f32b4fec487ec08.ppt