Генные болезни.ppt
- Количество слайдов: 113
 Доцент Чичко А. М. 1 -я кафедра детских болезней БГМУ
Доцент Чичко А. М. 1 -я кафедра детских болезней БГМУ
 Генные болезни - это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями единичных генов. В настоящее время описано более 3 тыс. наследственных болезней, обусловленных генными мутациями.
Генные болезни - это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями единичных генов. В настоящее время описано более 3 тыс. наследственных болезней, обусловленных генными мутациями.
 Генные мутации у человека являются причинами многих форм наследственной патологии. Основные из них: генные болезни, врожденные пороки развития и заболевания с наследственной
Генные мутации у человека являются причинами многих форм наследственной патологии. Основные из них: генные болезни, врожденные пороки развития и заболевания с наследственной
 У человека описаны следующие виды генных мутаций: мисценс, считывания, делеции, сплайсинга. нонсенс, инсерции, сдвиг рамки нарушения Одна и та же генная болезнь может быть обусловлена разными мутациями. Так, в гене муковисцидоза описано
У человека описаны следующие виды генных мутаций: мисценс, считывания, делеции, сплайсинга. нонсенс, инсерции, сдвиг рамки нарушения Одна и та же генная болезнь может быть обусловлена разными мутациями. Так, в гене муковисцидоза описано
 В некоторых случаях мутации в разных частях одного гена могут приводить к различным болезням (например, мутации RET-онкогена). Генные болезни наследуются по законам Менделя в том случае, когда речь идет о полных формах, обусловленных гаметическими мутациями. Это могут быть новые или унаследованные от предыдущих поколений мутации. В этих случаях патологические гены присутствуют во всех клетках организма.
В некоторых случаях мутации в разных частях одного гена могут приводить к различным болезням (например, мутации RET-онкогена). Генные болезни наследуются по законам Менделя в том случае, когда речь идет о полных формах, обусловленных гаметическими мутациями. Это могут быть новые или унаследованные от предыдущих поколений мутации. В этих случаях патологические гены присутствуют во всех клетках организма.
 Однако, генные мутации могут возникнуть в одной из клеток на разных стадиях дробления зиготы, и тогда организм будет мозаичен по данному гену. Если эта мутация доминантная, то она проявится фенотипически в соответствующих клетках и может приводить к развитию болезни (вероятно, менее тяжелой формы, чем у полных мутантов). Проблема мозаичных форм генных болезней и в генетическом, и в клиническом плане исследована недостаточно.
Однако, генные мутации могут возникнуть в одной из клеток на разных стадиях дробления зиготы, и тогда организм будет мозаичен по данному гену. Если эта мутация доминантная, то она проявится фенотипически в соответствующих клетках и может приводить к развитию болезни (вероятно, менее тяжелой формы, чем у полных мутантов). Проблема мозаичных форм генных болезней и в генетическом, и в клиническом плане исследована недостаточно.
 Тип болезни Доля общего числа, % Генные болезни Хромосомные Мультифакториальные Негенетические причины 8 -10 2 -3 35 -40 50
Тип болезни Доля общего числа, % Генные болезни Хромосомные Мультифакториальные Негенетические причины 8 -10 2 -3 35 -40 50
 Аутосомно-рецессивные (АР) Х- сцепленные доминантные Х- сцепленные рецессивные Y- сцепленные") Генетический принцип Аутосомно-доминантные (АД) Аутосомно-рецессивные (АР) Х- сцепленные доминантные Х- сцепленные рецессивные Y- сцепленные (голандрические) Митохондриальные
Генетический принцип Аутосомно-доминантные (АД) Аутосомно-рецессивные (АР) Х- сцепленные доминантные Х- сцепленные рецессивные Y- сцепленные (голандрические) Митохондриальные
 ВПР") Патогенетический принцип Наследственные болезни обмена веществ (углеводного, белкового, липидного, обмена витаминов, гормонов, металлов) ВПР Комбинированные состояния
Патогенетический принцип Наследственные болезни обмена веществ (углеводного, белкового, липидного, обмена витаминов, гормонов, металлов) ВПР Комбинированные состояния
 Клинический принцип Системные скелетные дисплазии Болезни с проявлениями гематологическими Нервно-мышечные болезни Болезни соединительной ткани Офтальмогенетика и др.
Клинический принцип Системные скелетные дисплазии Болезни с проявлениями гематологическими Нервно-мышечные болезни Болезни соединительной ткани Офтальмогенетика и др.
 Многообразие клинических проявлений Вариабельный возраст начала болезни Проградиентное хроническое течение Различная тяжесть проявлений, часто инвалидность Укорочение продолжительности жизни. Клинический полиморфизм заболеваний, антиципация Генетическая гетерогенность: межлокусная, внутрилокусная, генетический компаунд.
Многообразие клинических проявлений Вариабельный возраст начала болезни Проградиентное хроническое течение Различная тяжесть проявлений, часто инвалидность Укорочение продолжительности жизни. Клинический полиморфизм заболеваний, антиципация Генетическая гетерогенность: межлокусная, внутрилокусная, генетический компаунд.
 Манифестация. Патологические мутации могут реализовываться в разные периоды онтогенеза. Около 25% от всей генной патологии проявляется внутриутробно, в до пубертатном возрасте - 45%. 90% проявляется в пубертатном и юношеском возрасте. В возрасте старше 20 -и лет проявляется около 10% моногенных болезней.
Манифестация. Патологические мутации могут реализовываться в разные периоды онтогенеза. Около 25% от всей генной патологии проявляется внутриутробно, в до пубертатном возрасте - 45%. 90% проявляется в пубертатном и юношеском возрасте. В возрасте старше 20 -и лет проявляется около 10% моногенных болезней.
 Мутантный аллель патологический первичный продукт цепь биохимических процессов клетки органы организм.
Мутантный аллель патологический первичный продукт цепь биохимических процессов клетки органы организм.
 Первичные эффекты мутантных генов могут проявляться в 4 -х вариантах: отсутствие синтеза полипептида синтез аномального полипептида количественно недостаточный синтез -количественно избыточный синтез полипептида На основе первичного эффекта развертывается весь сложный патогенез генной болезни, проявляющийся определенной клинической картиной.
Первичные эффекты мутантных генов могут проявляться в 4 -х вариантах: отсутствие синтеза полипептида синтез аномального полипептида количественно недостаточный синтез -количественно избыточный синтез полипептида На основе первичного эффекта развертывается весь сложный патогенез генной болезни, проявляющийся определенной клинической картиной.
: Мукополисахаридозы (накопление гликозоаминогликанов) Гликогенозы (гликоген в") На клеточном уровне это проявляется болезнями накопления (лизосомные): Мукополисахаридозы (накопление гликозоаминогликанов) Гликогенозы (гликоген в клетках печени и мышц, нет расщепления до глюкозы) Пероксисомные болезни - 18 нозологических форм, проявляются: МВПР (черепно-лицевые дисморфии, катаракта, почечные кисты и др. )
На клеточном уровне это проявляется болезнями накопления (лизосомные): Мукополисахаридозы (накопление гликозоаминогликанов) Гликогенозы (гликоген в клетках печени и мышц, нет расщепления до глюкозы) Пероксисомные болезни - 18 нозологических форм, проявляются: МВПР (черепно-лицевые дисморфии, катаракта, почечные кисты и др. )
 Мембранными нарушениями – так, отсутствие рецептора на мембране, связывающего ЛП низкой плотности ведет к развитию семейной гиперхолестеринемии. Отсутствие или дефект рецепторов к 1, 25 дигидроксихолекальциферолу ведет к вит. Д резистентному рахиту (АД заболевание).
Мембранными нарушениями – так, отсутствие рецептора на мембране, связывающего ЛП низкой плотности ведет к развитию семейной гиперхолестеринемии. Отсутствие или дефект рецепторов к 1, 25 дигидроксихолекальциферолу ведет к вит. Д резистентному рахиту (АД заболевание).
.") Поражение печени и экстрапирамидной системы вследствие отложения ионов меди при гепатолентикулярной дегенерации (болезнь Вильсона-Коновалова).
Поражение печени и экстрапирамидной системы вследствие отложения ионов меди при гепатолентикулярной дегенерации (болезнь Вильсона-Коновалова).
 зависит") Тяжесть и скорость развития болезни прочих равных условиях (один пол, одинаковый характер мутации) зависит от генотипа организма (гены-модификаторы) и условий внешней среды. Патогенез любой болезни у разных индивидуумов, хотя и сходен по первичным механизмами этапам, но формируется индивидуально.
Тяжесть и скорость развития болезни прочих равных условиях (один пол, одинаковый характер мутации) зависит от генотипа организма (гены-модификаторы) и условий внешней среды. Патогенез любой болезни у разных индивидуумов, хотя и сходен по первичным механизмами этапам, но формируется индивидуально.
 Показаны при молекулярно-генетическом исследовании структуры генов, их мутаций и первичных продуктов. Так, мутации, приводящие к нарушению синтеза белка дистрофина, приводят к 2 -м клиническим формам: 1. Миопатии Дюшенна (тяжелая), при которой происходит полная блокада синтеза РНК для белка дистрофина. 2. Миопатия Беккера (легкая), при которой делеция гена по размеру меньше и происходит лишь частичная блокада синтеза белка.
Показаны при молекулярно-генетическом исследовании структуры генов, их мутаций и первичных продуктов. Так, мутации, приводящие к нарушению синтеза белка дистрофина, приводят к 2 -м клиническим формам: 1. Миопатии Дюшенна (тяжелая), при которой происходит полная блокада синтеза РНК для белка дистрофина. 2. Миопатия Беккера (легкая), при которой делеция гена по размеру меньше и происходит лишь частичная блокада синтеза белка.
 Сплайсинговые мутации не полностью блокируют образование матричной РНК и поэтому соответствующие формы болезни обычно легче по течению. Четкая корреляция обнаружена лишь при одном виде мутаций – экспансии тринуклеотидных повторов в гене. Экспансия формируется в мейозе у одного из родителей, это обуславливает явление антиципации – более тяжелое течение болезни в последующих поколениях, чем у родителей. Основа антиципации – то, что к деторождению предрасполагают более легкие формы болезни. Обнаружена при 6 болезнях.
Сплайсинговые мутации не полностью блокируют образование матричной РНК и поэтому соответствующие формы болезни обычно легче по течению. Четкая корреляция обнаружена лишь при одном виде мутаций – экспансии тринуклеотидных повторов в гене. Экспансия формируется в мейозе у одного из родителей, это обуславливает явление антиципации – более тяжелое течение болезни в последующих поколениях, чем у родителей. Основа антиципации – то, что к деторождению предрасполагают более легкие формы болезни. Обнаружена при 6 болезнях.
 Число триплетных повторов Болезнь Хро Повтоморяюсо- щийся ма трипле т Норма Премутация Болез Предп. пол нь Умственная отсталость с ломкой Ххромосомой (Х) Х CGG 6 -51 57 -200 200 -2000 Умственная отсталость с ломкой Ххромосомой ХЕ) Х GCC 6 -42 50 -200 М, Ж Болезнь Кеннеди Х CAG 11 -31 40 -62 М Хорея Гентингтона 4 р CAG 11 -34 30 -38 37 -86 М Спинномозжечковая 6 р CAG 25 -36 35 -43 42 -81 М Ж
Число триплетных повторов Болезнь Хро Повтоморяюсо- щийся ма трипле т Норма Премутация Болез Предп. пол нь Умственная отсталость с ломкой Ххромосомой (Х) Х CGG 6 -51 57 -200 200 -2000 Умственная отсталость с ломкой Ххромосомой ХЕ) Х GCC 6 -42 50 -200 М, Ж Болезнь Кеннеди Х CAG 11 -31 40 -62 М Хорея Гентингтона 4 р CAG 11 -34 30 -38 37 -86 М Спинномозжечковая 6 р CAG 25 -36 35 -43 42 -81 М Ж
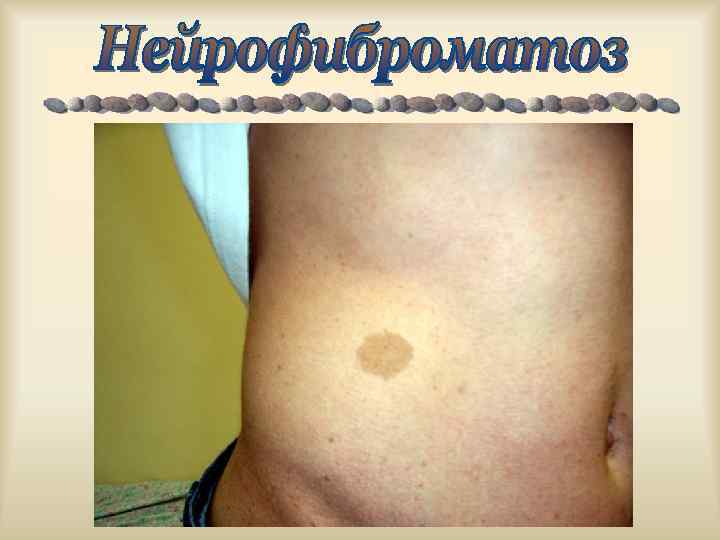
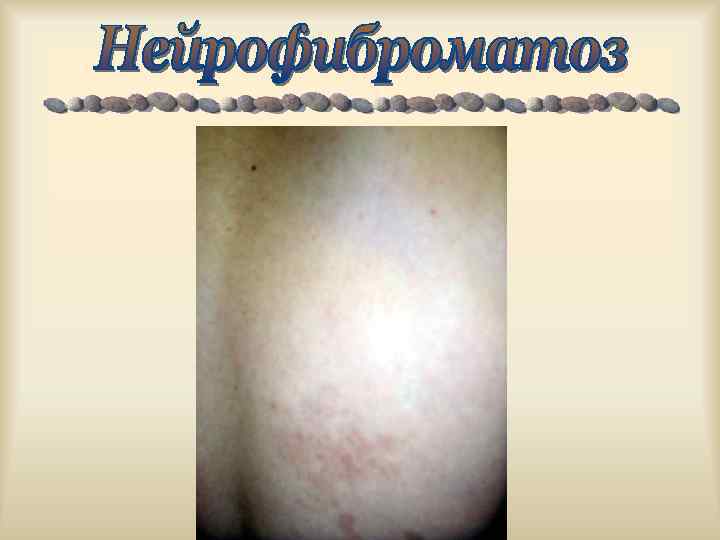
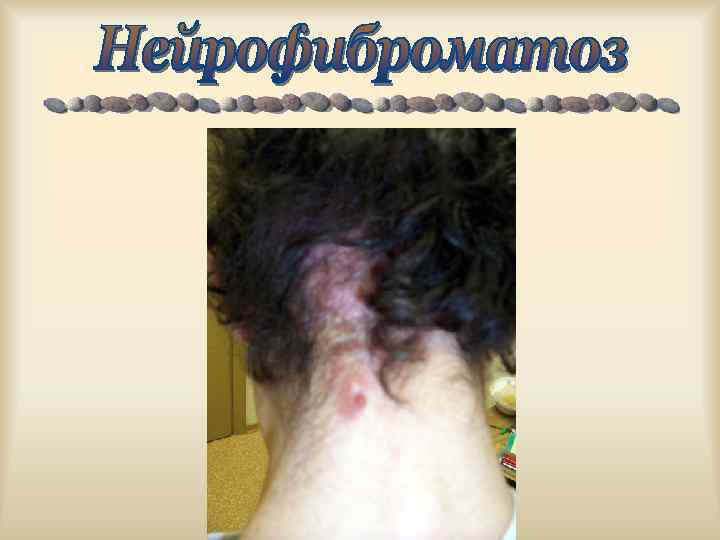
 Усиление симптомов ФКУ у ребенка в случае, если диета матери была богата продуктами с высоким содержанием ФА. Рост нейрофибром при нейрофиброматозе 1 у женщин резко усиливается при беременности. Обострение миодистрофий после стрессов, переутомлений, переохлаждений, инфекций.
Усиление симптомов ФКУ у ребенка в случае, если диета матери была богата продуктами с высоким содержанием ФА. Рост нейрофибром при нейрофиброматозе 1 у женщин резко усиливается при беременности. Обострение миодистрофий после стрессов, переутомлений, переохлаждений, инфекций.
 обусловлена мутациями в разных локусах или разными мутациями в одном локусе, т. е. один фенотип при разных нозологических формах.
обусловлена мутациями в разных локусах или разными мутациями в одном локусе, т. е. один фенотип при разных нозологических формах.
 при с. Элерса-Данло 11 форм, нейрофиброматозе - 6 форм, гликогенозы – более 6 форм; в этих группах имеются и АД, и АР, и Х-сц. формы, т. е. мутации в разных локусах, в разных хромосомах.
при с. Элерса-Данло 11 форм, нейрофиброматозе - 6 форм, гликогенозы – более 6 форм; в этих группах имеются и АД, и АР, и Х-сц. формы, т. е. мутации в разных локусах, в разных хромосомах.
 это сочетание 2 -х разных патологических аллелей одного локуса у индивида. Отличаются от гомозиготных форм по обеим аллелям.
это сочетание 2 -х разных патологических аллелей одного локуса у индивида. Отличаются от гомозиготных форм по обеим аллелям.
 Наиболее объективная оценка распростра-ненности в разных популяциях определение их частоты среди новорожденных, включая мертворожденных, так как затем в связи со смертностью, особенно в раннем возрасте, распространенность снижается. Смертность может различаться, т. к. зависит от многих факторов, в том числе и от уровня оказания медицинской помощи,
Наиболее объективная оценка распростра-ненности в разных популяциях определение их частоты среди новорожденных, включая мертворожденных, так как затем в связи со смертностью, особенно в раннем возрасте, распространенность снижается. Смертность может различаться, т. к. зависит от многих факторов, в том числе и от уровня оказания медицинской помощи,
 Генные болезни составляют в популяции около 1 % (АД- 0, 5%, АР- 0, 25%, Х-сц. - 0, 25%) Высокая, если заболевание встречается чаще, чем 1: 10. 000 Cредняя - 1: 10. 000 - 40. 000 Низкая - 1: 40. 000, 1: 100. 000 и реже. Распространенность 15 наиболее частых болезней обуславливает примерно 50% всей общей частоты.
Генные болезни составляют в популяции около 1 % (АД- 0, 5%, АР- 0, 25%, Х-сц. - 0, 25%) Высокая, если заболевание встречается чаще, чем 1: 10. 000 Cредняя - 1: 10. 000 - 40. 000 Низкая - 1: 40. 000, 1: 100. 000 и реже. Распространенность 15 наиболее частых болезней обуславливает примерно 50% всей общей частоты.
 Болезнь Распространенность Тип наследования Первичный гемохроматоз 1: 500 АР Неполипозный рак толстой кишки (синдром Линча) 1: 200 – 1: 2 000 АД Умственная недостаточность с ломкой Х -хромосомой (синдром Мартина-Белла) 1: 1 250 (мальчики) Муковисцидоз 1: 1 600 – 1: 3 000 АР Нейрофиброматоз 1: 4 000 АД Х-сцепленный 1: 2 500 (девочки)
Болезнь Распространенность Тип наследования Первичный гемохроматоз 1: 500 АР Неполипозный рак толстой кишки (синдром Линча) 1: 200 – 1: 2 000 АД Умственная недостаточность с ломкой Х -хромосомой (синдром Мартина-Белла) 1: 1 250 (мальчики) Муковисцидоз 1: 1 600 – 1: 3 000 АР Нейрофиброматоз 1: 4 000 АД Х-сцепленный 1: 2 500 (девочки)
 Болезнь Распространенность Тип наследования Спинальная мышечная атрофия 1: 6 000 АР Миотоническая дистрофия 1: 8 000 – 1: 10 000 АД Миопатия Дюшенна – Беккера 1: 3000– 1: 5000 (мальчики) Х – сцепленный рецессивный Синдром Элерса – Данло (все формы) 1: 5 000 А Д, А Р, Х- сцепленный Р Синдром Марфана 1: 10 000 – 1: 15 000 АД Фенилкетонурия 1: 10 000 АР Ахондроплазия 1: 100 000 АД
Болезнь Распространенность Тип наследования Спинальная мышечная атрофия 1: 6 000 АР Миотоническая дистрофия 1: 8 000 – 1: 10 000 АД Миопатия Дюшенна – Беккера 1: 3000– 1: 5000 (мальчики) Х – сцепленный рецессивный Синдром Элерса – Данло (все формы) 1: 5 000 А Д, А Р, Х- сцепленный Р Синдром Марфана 1: 10 000 – 1: 15 000 АД Фенилкетонурия 1: 10 000 АР Ахондроплазия 1: 100 000 АД
 Распространенность генных болезней сильно различается, нет зависимости от типа наследования. При АД типе наследования распространенность определяется новыми мутациями, т. к. наблюдается снижение фертильности, часто стерильность (так, при ахондроплазии только 10% семейные случаи). При поздно начинающихся болезнях – хореи Гентингтона, б. Альцгеймера - до клинических проявлений болезни деторождение как правило закончено, не отражается на количестве потомков.
Распространенность генных болезней сильно различается, нет зависимости от типа наследования. При АД типе наследования распространенность определяется новыми мутациями, т. к. наблюдается снижение фертильности, часто стерильность (так, при ахондроплазии только 10% семейные случаи). При поздно начинающихся болезнях – хореи Гентингтона, б. Альцгеймера - до клинических проявлений болезни деторождение как правило закончено, не отражается на количестве потомков.
 Распространенность АР болезней определяется частотой гетерозигот в популяциях. Происходит накопление гетерозигот в популяциях даже при отсутствии репродуктивного преимущества. Отягощенность грузом АР мутаций. Кровнородственные браки - маркер АР наследования.
Распространенность АР болезней определяется частотой гетерозигот в популяциях. Происходит накопление гетерозигот в популяциях даже при отсутствии репродуктивного преимущества. Отягощенность грузом АР мутаций. Кровнородственные браки - маркер АР наследования.
 1% на уровне двоюродных братьев и сестер в Европе (до 10 -20% в ряде этнических групп) ведет к росту гомозигот по рецессивным генам. Так, альбинизм встречается у потомства неродственных браков - 1: 40. 000, и 1: 3000 - у двоюродных. Ихтиозников, : 1. 000 у 1: 16. 000 - родственников.
1% на уровне двоюродных братьев и сестер в Европе (до 10 -20% в ряде этнических групп) ведет к росту гомозигот по рецессивным генам. Так, альбинизм встречается у потомства неродственных браков - 1: 40. 000, и 1: 3000 - у двоюродных. Ихтиозников, : 1. 000 у 1: 16. 000 - родственников.
 в регионах с высокой") Распространенность АРгемоглобинопатий (серповидно-клеточная анемия, талассемия, аномальные гемоглобины и др. ) в регионах с высокой заболеваемостью малярией (Юго-восточная Азия, Африка, Италия, Азербайджан и др. ). Влияет и низкая репродукция – при гемофилии, ахондроплазии, нейрофиброматозе 1. Однако, часто количество детей в семьях с наследственной патологией больше,
Распространенность АРгемоглобинопатий (серповидно-клеточная анемия, талассемия, аномальные гемоглобины и др. ) в регионах с высокой заболеваемостью малярией (Юго-восточная Азия, Африка, Италия, Азербайджан и др. ). Влияет и низкая репродукция – при гемофилии, ахондроплазии, нейрофиброматозе 1. Однако, часто количество детей в семьях с наследственной патологией больше,
 Играет роль и увеличение продолжительности жизни больных с ФКУ, муковисцидозом, больные могут вступать в брак, иметь детей, что ведет к увеличению гетерозигот в популяции. Имеет значение дрейф генов (повышение частоты какого либо аллеля вследствие случайных событий, особенно в изоляте (5001500 человек).
Играет роль и увеличение продолжительности жизни больных с ФКУ, муковисцидозом, больные могут вступать в брак, иметь детей, что ведет к увеличению гетерозигот в популяции. Имеет значение дрейф генов (повышение частоты какого либо аллеля вследствие случайных событий, особенно в изоляте (5001500 человек).
 Эффект родоначальника – так, миграция в ЮАР выходцев из Голландии, Дании, Германии привело к значительной распространенности в ЮАР среди белых порфирии (АД), хореи Гентингтона, семейного полипоза толстой кишки (АД), липопротеиноза(АР) и др. что привело к большей частоте болезней в ЮАР, чем в других странах.
Эффект родоначальника – так, миграция в ЮАР выходцев из Голландии, Дании, Германии привело к значительной распространенности в ЮАР среди белых порфирии (АД), хореи Гентингтона, семейного полипоза толстой кишки (АД), липопротеиноза(АР) и др. что привело к большей частоте болезней в ЮАР, чем в других странах.
 Редкие болезни относительно часто встречаются в ряде популяций. Так, у евреев ашкенази (США, Европа, Израиль) часты болезни накопления – б. Тея. Сакса, Нимана-Пика, Гоше, а-беталипопротеинемия, брахидактилия и др. , Финны чаще страдают врожденным нефрозом Финского типа, липофусцинозом Армяне - семейной средиземноморской лихорадкой (периодическая болезнь).
Редкие болезни относительно часто встречаются в ряде популяций. Так, у евреев ашкенази (США, Европа, Израиль) часты болезни накопления – б. Тея. Сакса, Нимана-Пика, Гоше, а-беталипопротеинемия, брахидактилия и др. , Финны чаще страдают врожденным нефрозом Финского типа, липофусцинозом Армяне - семейной средиземноморской лихорадкой (периодическая болезнь).
 с. Элерса-Данло, хорея Гентингтона, гемофилия. Канадцы французского происхождения тирозинемия, Тея-Сакса,") Азербайджанцы - (отдельные районы) с. Элерса-Данло, хорея Гентингтона, гемофилия. Канадцы французского происхождения тирозинемия, Тея-Сакса, Афроамериканцы - серповидно-клеточная анемия, Европейцы и выходцы из Европы чаще страдают муковисцидозом. Таласемией, Греки, итальянцы - бета-Таласемия. Как правило, это связано с брачной изоляцией, даже вне географических ограничений.
Азербайджанцы - (отдельные районы) с. Элерса-Данло, хорея Гентингтона, гемофилия. Канадцы французского происхождения тирозинемия, Тея-Сакса, Афроамериканцы - серповидно-клеточная анемия, Европейцы и выходцы из Европы чаще страдают муковисцидозом. Таласемией, Греки, итальянцы - бета-Таласемия. Как правило, это связано с брачной изоляцией, даже вне географических ограничений.
 1. Заболевание передается из поколения в поколение, каждый больной ребенок имеет больного родителя (наследование по вертикали) 2. Так как патологический ген локализуется в аутосоме, одинаково часто болеют и мальчики, и девочки. 3. Нормальные дети больных родителей имеют нормальных всех детей. 4. Гомозиготы по патологическому гену рождаются от двух больных родителей, болезнь у них протекает тяжелее 5. Вероятность рождения больного ребенка, если болен один из родителей - 50%
1. Заболевание передается из поколения в поколение, каждый больной ребенок имеет больного родителя (наследование по вертикали) 2. Так как патологический ген локализуется в аутосоме, одинаково часто болеют и мальчики, и девочки. 3. Нормальные дети больных родителей имеют нормальных всех детей. 4. Гомозиготы по патологическому гену рождаются от двух больных родителей, болезнь у них протекает тяжелее 5. Вероятность рождения больного ребенка, если болен один из родителей - 50%
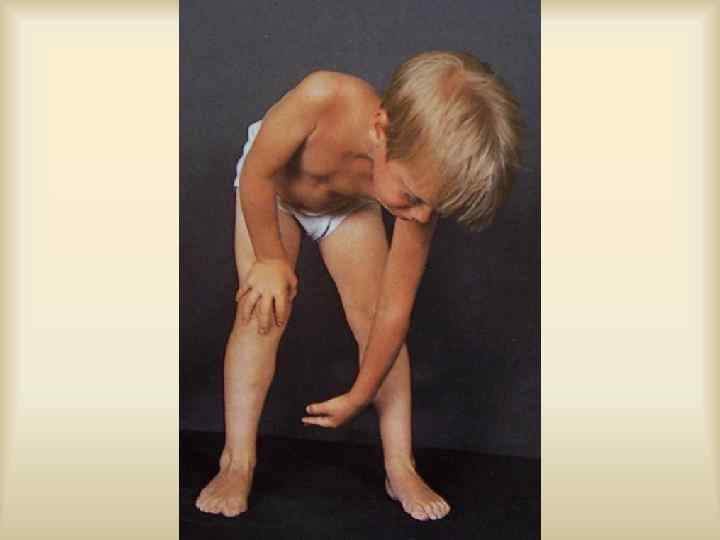
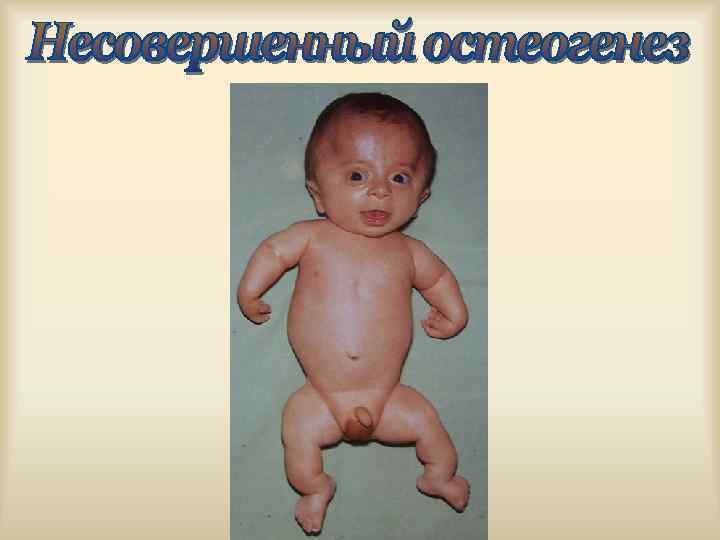
,") Характерен клинический полиморфизм: пенетрантность, экспрессивность генов. Ахондроплазия, с. Альпорта, с. Марфана, Элерса-Данлоса, Реклингаузена, Бурневиля), (микрофтальм), нейрофиброматоз туберозный склероз изолированные АДПП, ( с. ВПР несовершенный остеогенез, с. Пфайфера, хорея Гентингтона.
Характерен клинический полиморфизм: пенетрантность, экспрессивность генов. Ахондроплазия, с. Альпорта, с. Марфана, Элерса-Данлоса, Реклингаузена, Бурневиля), (микрофтальм), нейрофиброматоз туберозный склероз изолированные АДПП, ( с. ВПР несовершенный остеогенез, с. Пфайфера, хорея Гентингтона.
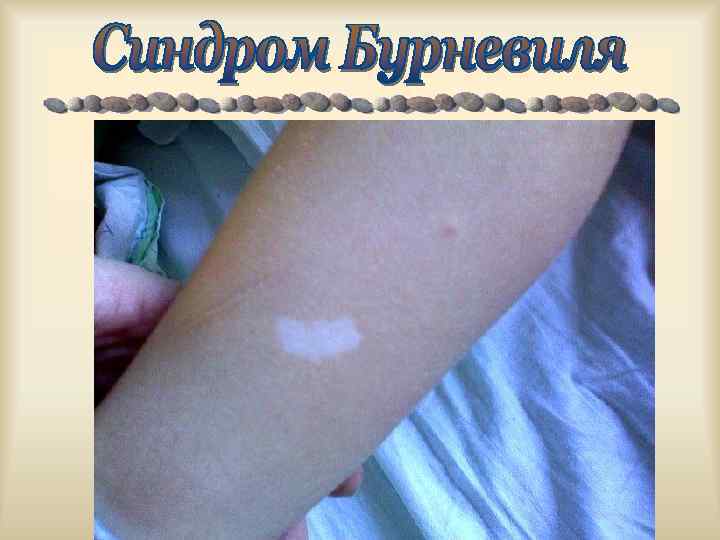
 Болеют в основном сибсы - дети фенотипически здоровых родителей, гетерозиготных носителей патологического гена (наследование по горизонтали). Болеют только гомозиготные носители гена, гетерозиготы здоровы. 1. 2. Часты кровнородственные родословной (маркер). браки в 3. Поражается 25% потомства (в случае гетерозиготного носительства родителями гена).
Болеют в основном сибсы - дети фенотипически здоровых родителей, гетерозиготных носителей патологического гена (наследование по горизонтали). Болеют только гомозиготные носители гена, гетерозиготы здоровы. 1. 2. Часты кровнородственные родословной (маркер). браки в 3. Поражается 25% потомства (в случае гетерозиготного носительства родителями гена).
 4. Если больны оба супруга, то все дети будут больны. 5. Оба пола поражаются с одинаковой частотой. 6. Больной индивидуум обычно имеет здоровое потомство (но увеличивает частоту гетерозигот в популяции!). Характерны энзимопатии: ФКУ, галактоземия, альбинизм, АРПП, АГС, муковисцидоз, мукополисахаридозы.
4. Если больны оба супруга, то все дети будут больны. 5. Оба пола поражаются с одинаковой частотой. 6. Больной индивидуум обычно имеет здоровое потомство (но увеличивает частоту гетерозигот в популяции!). Характерны энзимопатии: ФКУ, галактоземия, альбинизм, АРПП, АГС, муковисцидоз, мукополисахаридозы.
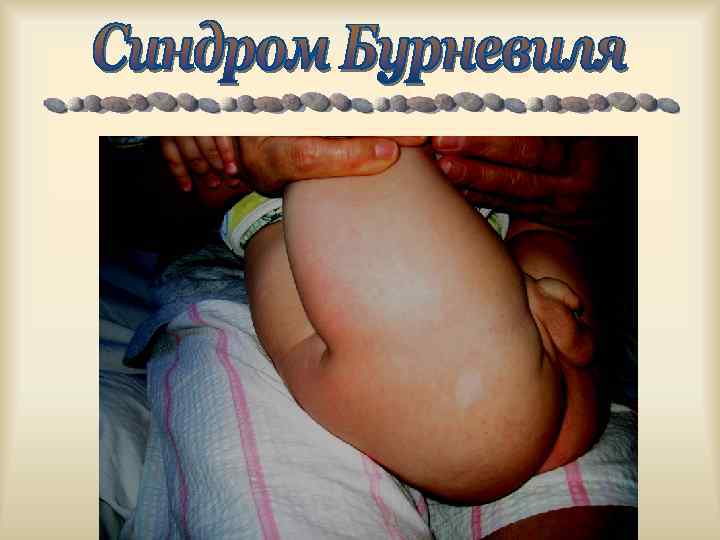
 Эхо. КГ от 15. 11. 05. , заключение: Рабдомиома правого желудочка без гемодинамических нарушений. Размеры камер сердца соответствуют физическому развитию. Общая сократительная способность миокарда левого желудочка удовлетворительная.
Эхо. КГ от 15. 11. 05. , заключение: Рабдомиома правого желудочка без гемодинамических нарушений. Размеры камер сердца соответствуют физическому развитию. Общая сократительная способность миокарда левого желудочка удовлетворительная.
 1. Заболевание наблюдается исключительно у мужчин. 2. Женщины гетерозиготны, являются носителями, фенотипически здоровы. 3. Риск рождения больного сына у женщины-носительницы - 50%, риск рождения дочери-носительницы мутантного гена - 50%.
1. Заболевание наблюдается исключительно у мужчин. 2. Женщины гетерозиготны, являются носителями, фенотипически здоровы. 3. Риск рождения больного сына у женщины-носительницы - 50%, риск рождения дочери-носительницы мутантного гена - 50%.
 4. У больного отца все сыновья здоровы, все дочери клинически здоровы, но являются облигатными носительницами. 5. Сын никогда не наследует заболевание отца. У носительниц патологического гена могут быть минимальные клинические проявления. Примерно в 1/3 случаев Х-сцепленная патология у мальчика может быть следствием новой мутации, а не наследуется от матери. Гемофилия А и В, мышечная дистрофия
4. У больного отца все сыновья здоровы, все дочери клинически здоровы, но являются облигатными носительницами. 5. Сын никогда не наследует заболевание отца. У носительниц патологического гена могут быть минимальные клинические проявления. Примерно в 1/3 случаев Х-сцепленная патология у мальчика может быть следствием новой мутации, а не наследуется от матери. Гемофилия А и В, мышечная дистрофия
, либо выражены различия") Болеют либо только девочки (гемизиготизация приводит к гибели эмбрионов мужского пола), либо выражены различия в тяжести клинических проявлений болезни в зависимости от пола – тяжело больны именно мальчики.
Болеют либо только девочки (гемизиготизация приводит к гибели эмбрионов мужского пола), либо выражены различия в тяжести клинических проявлений болезни в зависимости от пола – тяжело больны именно мальчики.
 1 -й вариант - болеют лица мужского и женского пола, но больных девочек в 2 раза больше, т. к. мальчики погибают внутриутробно, выжившие тяжело болеют. От больного отца заболевание передается всем дочерям, но не передается сыновьям. От больной матери заболевание передается с одинаковой вероятностью 50% дочерей и 50% сыновей (как при АД типе наследования). Фосфат-диабет или Витамин Д – резистентный рахит.
1 -й вариант - болеют лица мужского и женского пола, но больных девочек в 2 раза больше, т. к. мальчики погибают внутриутробно, выжившие тяжело болеют. От больного отца заболевание передается всем дочерям, но не передается сыновьям. От больной матери заболевание передается с одинаковой вероятностью 50% дочерей и 50% сыновей (как при АД типе наследования). Фосфат-диабет или Витамин Д – резистентный рахит.
 2 -й вариант - мальчики не рождаются, болеют только женщины, при этом они имеют в родословной аборты повторные вследствие гибели спонтанные эмбрионов мужского пола на ранних стадиях развития.
2 -й вариант - мальчики не рождаются, болеют только женщины, при этом они имеют в родословной аборты повторные вследствие гибели спонтанные эмбрионов мужского пола на ранних стадиях развития.
 Передается от отца всем мальчикам. Оволосение ушной раковины. Клинического значения не имеет.
Передается от отца всем мальчикам. Оволосение ушной раковины. Клинического значения не имеет.
. 1. Болезнь") Передаются с цитоплазмой ооцитов (кольцевая хромосома митохондрии, > 16 тыс. пар оснований). 1. Болезнь передается только от матери, 2. Больны и мальчики и девочки. 3. Больные отцы не передают болезни ни дочерям, ни сыновьям. Атрофии зрительного нерва, прогрессирующие офтальмоплегии.
Передаются с цитоплазмой ооцитов (кольцевая хромосома митохондрии, > 16 тыс. пар оснований). 1. Болезнь передается только от матери, 2. Больны и мальчики и девочки. 3. Больные отцы не передают болезни ни дочерям, ни сыновьям. Атрофии зрительного нерва, прогрессирующие офтальмоплегии.
 Частота 1: 6 000 -10 000, в Беларуси -1: 6 000, носитель мутации") Фенилкетонурия(ФКУ) Частота 1: 6 000 -10 000, в Беларуси -1: 6 000, носитель мутации гена каждый 48 -й. Наследуется по аутосомно-рецессивному типу; больные - рецессивные гомозиготы (аа). Мутантный ген, который отвечает за синтез фермента фенилаланингидроксилазы, картирован (12 q 22 -q 24), идентифицирован и секвенирован (определена последовательность нуклеотидов).
Фенилкетонурия(ФКУ) Частота 1: 6 000 -10 000, в Беларуси -1: 6 000, носитель мутации гена каждый 48 -й. Наследуется по аутосомно-рецессивному типу; больные - рецессивные гомозиготы (аа). Мутантный ген, который отвечает за синтез фермента фенилаланингидроксилазы, картирован (12 q 22 -q 24), идентифицирован и секвенирован (определена последовательность нуклеотидов).
 70% в РБ мутация (замена R 408 W аминокислоты аргинина на триптофан в 408 положении. Фенилаланин принадлежит к числу незаменимых аминокислот. Только часть ФА используется для синтеза белков; основное количество этой аминокислоты окисляется до тирозина.
70% в РБ мутация (замена R 408 W аминокислоты аргинина на триптофан в 408 положении. Фенилаланин принадлежит к числу незаменимых аминокислот. Только часть ФА используется для синтеза белков; основное количество этой аминокислоты окисляется до тирозина.
 Если фермент фенилаланингидроксилаза не активен, то ФА не превращается в тирозин, а накапливается в сыворотке крови в больших количествах в виде фенилпировиноградной кислоты (ФПВК), которая выделяется с мочой и потом, вследствие чего от больных исходит "мышиный" запах. Высокая концентрация ФПВК приводит к нарушению формирования миелиновой оболочки
Если фермент фенилаланингидроксилаза не активен, то ФА не превращается в тирозин, а накапливается в сыворотке крови в больших количествах в виде фенилпировиноградной кислоты (ФПВК), которая выделяется с мочой и потом, вследствие чего от больных исходит "мышиный" запах. Высокая концентрация ФПВК приводит к нарушению формирования миелиновой оболочки
 Дети с ФКУ рождаются здоровыми, но в первые же недели жизни у них развиваются клинические проявления. ФПВК является нейротропным ядом, в результате чего повышаются возбудимость, тонус мышц, развиваются гиперрефлексия, тремор, судорожные эпилептиформные припадки.
Дети с ФКУ рождаются здоровыми, но в первые же недели жизни у них развиваются клинические проявления. ФПВК является нейротропным ядом, в результате чего повышаются возбудимость, тонус мышц, развиваются гиперрефлексия, тремор, судорожные эпилептиформные припадки.
 Позже присоединяются нарушения высшей нервной деятельности, умственная отсталость, пигментация меланина. микроцефалия. из-за нарушения синтеза
Позже присоединяются нарушения высшей нервной деятельности, умственная отсталость, пигментация меланина. микроцефалия. из-за нарушения синтеза
 Фенилкетонурия I аутосомно-рецессивным типом наследования, вызываемое мутациями гена РАН, локализующееся на длинном плече 12 -й хромосомы (12 q 24. 1). Фенилкетонурия II также наследуется по аутосомно-рецессивному типу, генный дефект локализуется в коротком плече 4 -й хромосомы, участке 4 p 15. 3. частота заболевания 1: 100 000.
Фенилкетонурия I аутосомно-рецессивным типом наследования, вызываемое мутациями гена РАН, локализующееся на длинном плече 12 -й хромосомы (12 q 24. 1). Фенилкетонурия II также наследуется по аутосомно-рецессивному типу, генный дефект локализуется в коротком плече 4 -й хромосомы, участке 4 p 15. 3. частота заболевания 1: 100 000.
 Вследствие недостаточности дигидроптеридинредуктазы нарушается восстановление активной формы тетрагидробиоптерина, участвующего в качестве кофактора в гидроксилировании фенилаланина, тирозина и триптофана, что приводит к накоплению метаболитов, нарушению образования предшественников нейромедиаторов катехоламинового и серотонинового ряда. В патогенезе заболевания имеет значение также снижение уровня фолатов в сыворотке крови, эритроцитах, спиномозговой жидкости.
Вследствие недостаточности дигидроптеридинредуктазы нарушается восстановление активной формы тетрагидробиоптерина, участвующего в качестве кофактора в гидроксилировании фенилаланина, тирозина и триптофана, что приводит к накоплению метаболитов, нарушению образования предшественников нейромедиаторов катехоламинового и серотонинового ряда. В патогенезе заболевания имеет значение также снижение уровня фолатов в сыворотке крови, эритроцитах, спиномозговой жидкости.
 Фенилкетонурия III наследуется по аутосомно-рецессивному типу и связано с недостаточностью 6 -пирувоилтетрагидроптерина синтазы, которая участвует в синтезе тетрагидробиоптерина из дигидронеоптерина трифосфата. Частота заболевания составляет 1: 30000. Главную роль в генезе заболевания играет дефицит тетрагидробиоптерина.
Фенилкетонурия III наследуется по аутосомно-рецессивному типу и связано с недостаточностью 6 -пирувоилтетрагидроптерина синтазы, которая участвует в синтезе тетрагидробиоптерина из дигидронеоптерина трифосфата. Частота заболевания составляет 1: 30000. Главную роль в генезе заболевания играет дефицит тетрагидробиоптерина.
 осуществляется биохимическими методами: в моче определяется ФПВК, в крови - высокое содержание фенилаланина (тонкослойная хроматография аминокислот). С 1978 года в РБ на 4 -й день жизни новорожденного проводится скрининг новорожденных на ФКУ. Проба крови отправляется в МГЦ, до 2, 5 мг% N (при белковой нагрузке 100 мг ФА в день). М. б. гипер. ФАемия до 10 мг%, более 10 мг% - ФКУ.
осуществляется биохимическими методами: в моче определяется ФПВК, в крови - высокое содержание фенилаланина (тонкослойная хроматография аминокислот). С 1978 года в РБ на 4 -й день жизни новорожденного проводится скрининг новорожденных на ФКУ. Проба крови отправляется в МГЦ, до 2, 5 мг% N (при белковой нагрузке 100 мг ФА в день). М. б. гипер. ФАемия до 10 мг%, более 10 мг% - ФКУ.
 Эффективным методом является диетотерапия - кормление ребенка пищей с низким содержанием фенилаланина. Для предотвращения необратимых поражений мозга лечение необходимо начинать с первых недель жизни и постоянно следить за содержанием ФА в крови.
Эффективным методом является диетотерапия - кормление ребенка пищей с низким содержанием фенилаланина. Для предотвращения необратимых поражений мозга лечение необходимо начинать с первых недель жизни и постоянно следить за содержанием ФА в крови.
 для грудных детей (ХР –") Ограничивается естественный белок и добавляются белковые препараты (смеси аминокислот) для грудных детей (ХР – Англ. + грудное молоко) РАМ-2 (старше года), Тетрафен, берлофен, лофеналак.
Ограничивается естественный белок и добавляются белковые препараты (смеси аминокислот) для грудных детей (ХР – Англ. + грудное молоко) РАМ-2 (старше года), Тетрафен, берлофен, лофеналак.
. Индивидуальный контроль питания в связи") Ограничиваются хлеб, мясо, яйцо, рыбу, можно овощи (ограничивается картофель). Индивидуальный контроль питания в связи с индивидуальной переносимостью ФА. ! Норма -2 -6 мг%, контролируется в 1 -й год жизни 1 раз в 1 неделю. Пожизненный контроль, особенно пока учится, хотя навыки не утрачиваются без
Ограничиваются хлеб, мясо, яйцо, рыбу, можно овощи (ограничивается картофель). Индивидуальный контроль питания в связи с индивидуальной переносимостью ФА. ! Норма -2 -6 мг%, контролируется в 1 -й год жизни 1 раз в 1 неделю. Пожизненный контроль, особенно пока учится, хотя навыки не утрачиваются без
 Проблемы – при планировании детей – строгое соблюдение диеты за 3 мес. до и всю беременность (РАМ - матерна). Риск гипотрофии, микроцефалии, ВПС плода (синдром материнской ФКУ). Умственная отсталость не прямо пропорциональна уровню ФА, больше зависит от проницаемости ГЭ барьера в ЦНС.
Проблемы – при планировании детей – строгое соблюдение диеты за 3 мес. до и всю беременность (РАМ - матерна). Риск гипотрофии, микроцефалии, ВПС плода (синдром материнской ФКУ). Умственная отсталость не прямо пропорциональна уровню ФА, больше зависит от проницаемости ГЭ барьера в ЦНС.
, диагностика США – семьи, генная терапия") Дородовая диагностика: биопсия ворсин хориона (с 11 недель), диагностика США – семьи, генная терапия ДНК ребенка. на мышах. Развитые страны: не стоит прерывать беременность, так как эффективна диета, государство оплачивает питание.
Дородовая диагностика: биопсия ворсин хориона (с 11 недель), диагностика США – семьи, генная терапия ДНК ребенка. на мышах. Развитые страны: не стоит прерывать беременность, так как эффективна диета, государство оплачивает питание.
. В РБ - 1:") Популяционная частота - 1: 2 500 (в странах северной Европы). В РБ - 1: 8150. У Африканцев, Японцев-1: 100. 000. Обусловлен мутацией в 7 -й хромосоме (7 q 31 -q 32), его величина - 250 000 пар нуклеотидов. Тип наследования - аутосомно-рецессивный. В настоящее время насчитывается более 800 мутаций в гене муковисцидоза, однако лишь 6 из них (del. F 508, G 542 X, G 551 D, R 553 X, W 1282 X, N 1303 K) в странах Европы встречаются с частотой более 1%. Одной из наиболее частых является мутация del 508 F, в Европе и РБ до 70%, частота в России составляет 56%.
Популяционная частота - 1: 2 500 (в странах северной Европы). В РБ - 1: 8150. У Африканцев, Японцев-1: 100. 000. Обусловлен мутацией в 7 -й хромосоме (7 q 31 -q 32), его величина - 250 000 пар нуклеотидов. Тип наследования - аутосомно-рецессивный. В настоящее время насчитывается более 800 мутаций в гене муковисцидоза, однако лишь 6 из них (del. F 508, G 542 X, G 551 D, R 553 X, W 1282 X, N 1303 K) в странах Европы встречаются с частотой более 1%. Одной из наиболее частых является мутация del 508 F, в Европе и РБ до 70%, частота в России составляет 56%.
 лежит нарушение транспорта ионов Cl- и Na+ через мембраны") В основе патогенеза муковисцидоза (МВ) лежит нарушение транспорта ионов Cl- и Na+ через мембраны эпителиальных клеток, вследствие отсутствия или изменения структуры белка трансмембранного регулятора проводимости (CFTR). В настоящее время считают, что этот белок является собственно хлорным каналом. Это приводит к избыточному выведению хлоридов, следствием чего является гиперсекреция густой слизи в клетках экзокринной части поджелудочной железы, эпителии бронхов, слизистой оболочки желудочно-кишечного тракта.
В основе патогенеза муковисцидоза (МВ) лежит нарушение транспорта ионов Cl- и Na+ через мембраны эпителиальных клеток, вследствие отсутствия или изменения структуры белка трансмембранного регулятора проводимости (CFTR). В настоящее время считают, что этот белок является собственно хлорным каналом. Это приводит к избыточному выведению хлоридов, следствием чего является гиперсекреция густой слизи в клетках экзокринной части поджелудочной железы, эпителии бронхов, слизистой оболочки желудочно-кишечного тракта.
 Муковисцидоз представляет собой множествен-ные поражения желез внешней секреции, проявляющиеся выделением секретов повышенной вязкости, что ведет к застойным явлениям и закупорке протоков в соответствующих органах (легких, поджелудочной железе и кишечнике) с последующими воспалительными процессами и склеротическими изменениями. В потовой жидкости повышена концентрация ионов Na+ и Cl-, что является основным диагностическим лабораторным тестом.
Муковисцидоз представляет собой множествен-ные поражения желез внешней секреции, проявляющиеся выделением секретов повышенной вязкости, что ведет к застойным явлениям и закупорке протоков в соответствующих органах (легких, поджелудочной железе и кишечнике) с последующими воспалительными процессами и склеротическими изменениями. В потовой жидкости повышена концентрация ионов Na+ и Cl-, что является основным диагностическим лабораторным тестом.
 Заболевание может манифестировать в любом возрасте.
Заболевание может манифестировать в любом возрасте.
 Гнилостные процессы в кишечнике приводят к вздутию живота, появлению обильного жирного стула с резким гнилостным запахом, нейтральному жиру в кале, отставанию в физическом развитии, гипотрофии, выпадению прямой кишки. В некоторых случаях развивается билиарный цирроз печени при поражении желчных протоков (печеночная форма).
Гнилостные процессы в кишечнике приводят к вздутию живота, появлению обильного жирного стула с резким гнилостным запахом, нейтральному жиру в кале, отставанию в физическом развитии, гипотрофии, выпадению прямой кишки. В некоторых случаях развивается билиарный цирроз печени при поражении желчных протоков (печеночная форма).
 связана с поражением бронхо-легочных желез с развитием обструкции дыхательных путей,") Преимущественно легочная форма (1520%) связана с поражением бронхо-легочных желез с развитием обструкции дыхательных путей, хронических форм воспалительного процесса с развитием эмфиземы и фиброза легких, характерны полипы и синуситы. Указанные проявления возникают уже на 1 -3 году жизни.
Преимущественно легочная форма (1520%) связана с поражением бронхо-легочных желез с развитием обструкции дыхательных путей, хронических форм воспалительного процесса с развитием эмфиземы и фиброза легких, характерны полипы и синуситы. Указанные проявления возникают уже на 1 -3 году жизни.
 смешанная форма муковисцидоза - (легочно-кишечная). При этой возникают форме на") Наиболее частая (75 -80%) смешанная форма муковисцидоза - (легочно-кишечная). При этой возникают форме на легочные 1 -2 -м проявления году характеризуются жизни и рецидивирующими пневмониями и бронхитами с последующим развитием эмфиземы недостаточности, а кишечные проявления. легких также и легочной наблюдаются и
Наиболее частая (75 -80%) смешанная форма муковисцидоза - (легочно-кишечная). При этой возникают форме на легочные 1 -2 -м проявления году характеризуются жизни и рецидивирующими пневмониями и бронхитами с последующим развитием эмфиземы недостаточности, а кишечные проявления. легких также и легочной наблюдаются и
 Реже встречаются абортивные и стертые формы - бесплодие у взрослых вследствие обструкции семенных канальцев у лиц мужского пола и густой шеечной слизи у женщин. Обычно у гетерозигот по мутантным генам. Важен семейный анамнез + потовая проба. У всех больных отмечается липкий соленый пот.
Реже встречаются абортивные и стертые формы - бесплодие у взрослых вследствие обструкции семенных канальцев у лиц мужского пола и густой шеечной слизи у женщин. Обычно у гетерозигот по мутантным генам. Важен семейный анамнез + потовая проба. У всех больных отмечается липкий соленый пот.
 Основана на клинической картине, определении натрия и хлоридов в потовом тесте (положителен при уровне Na и Cl более 60 сомнителен (разбежка не более 20), - 40 -60, нормально до 40 ммоль/л. Тест может надпочечниковой быть ложно + при недостаточности, гипотиреозе, гипопаратиреозе, несахарном диабете, мукополисахаридозе и др.
Основана на клинической картине, определении натрия и хлоридов в потовом тесте (положителен при уровне Na и Cl более 60 сомнителен (разбежка не более 20), - 40 -60, нормально до 40 ммоль/л. Тест может надпочечниковой быть ложно + при недостаточности, гипотиреозе, гипопаратиреозе, несахарном диабете, мукополисахаридозе и др.
 Молекулярно-генетическая диагностика определение мутации гена CFTR del 508 F, в том числе пренатально. Скрининг новорожденных – определение иммунореактивного трипсина в крови.
Молекулярно-генетическая диагностика определение мутации гена CFTR del 508 F, в том числе пренатально. Скрининг новорожденных – определение иммунореактивного трипсина в крови.
 Критерии диагностики: Типичные легочные и/или гастроинтестинальные проявления. Наличие пораженных сибсов в семье, Потовая проба более 60 ммоль/л (после 1 мес. жизни), Выявление мутаций в 2 -х хромосомах. Диагноз достоверен при сочетании любого из клинических критериев и 3 или 4.
Критерии диагностики: Типичные легочные и/или гастроинтестинальные проявления. Наличие пораженных сибсов в семье, Потовая проба более 60 ммоль/л (после 1 мес. жизни), Выявление мутаций в 2 -х хромосомах. Диагноз достоверен при сочетании любого из клинических критериев и 3 или 4.
 Улучшился в последние годы. 10 -20% детей с МВ погибают до 1 года жизни, до 30 лет 50%. За рубежом единичные случаи продолжительности жизни до 60 лет.
Улучшился в последние годы. 10 -20% детей с МВ погибают до 1 года жизни, до 30 лет 50%. За рубежом единичные случаи продолжительности жизни до 60 лет.
") 1. Антибактериальная терапия при наличии хронического воспалительного процесса в БЛС (чаще вызывается синегнойной палочкой) 2 -3 антибиотика, не менее 1 мес. , высокие дозы пролонгированные курсы. Фторхинолоны, аминогликозиды, цефалоспорины. Профилактическое применение в виде ингаляций. 2. Ферментотерапия даже при легких формах – в р. Н чувствительной оболочке, гранулированные, с высокими дозами липазы, амилазы, протеазы (креон, панцитрат) до нормализации стула визуально, копрограммы, набора веса.
1. Антибактериальная терапия при наличии хронического воспалительного процесса в БЛС (чаще вызывается синегнойной палочкой) 2 -3 антибиотика, не менее 1 мес. , высокие дозы пролонгированные курсы. Фторхинолоны, аминогликозиды, цефалоспорины. Профилактическое применение в виде ингаляций. 2. Ферментотерапия даже при легких формах – в р. Н чувствительной оболочке, гранулированные, с высокими дозами липазы, амилазы, протеазы (креон, панцитрат) до нормализации стула визуально, копрограммы, набора веса.
 3. Диета – дополнительно соль, нет ограничений, контролируется ферментами. Водный режим – около 4 литров в сутки. 4. Муколитики внутрь – рекомбинантная ДНКаза (Пульмозим) и др. , ежедневные 2 -3 р. ингаляции, постуральный дренаж, флаттертерапия, дыхание с положительным давлением на выдохе и др. 5. Витаминотерапия, особенно жирорастворимые, 2 -3 р. в день. Генная терапия - аэрозоли с геном МВ. Трансплантация легких, сердца удлиняет примерно на 10 лет жизнь.
3. Диета – дополнительно соль, нет ограничений, контролируется ферментами. Водный режим – около 4 литров в сутки. 4. Муколитики внутрь – рекомбинантная ДНКаза (Пульмозим) и др. , ежедневные 2 -3 р. ингаляции, постуральный дренаж, флаттертерапия, дыхание с положительным давлением на выдохе и др. 5. Витаминотерапия, особенно жирорастворимые, 2 -3 р. в день. Генная терапия - аэрозоли с геном МВ. Трансплантация легких, сердца удлиняет примерно на 10 лет жизнь.


 Умственная отсталость с ломкой Х- хромосомой, FRA-X синдром. Наиболее частая после болезни Дауна умственная отсталость, 1: 2000 -2500 новорожденных, мальчиков в 2 -3 раза больше, болеют тяжелее. Признаки: Умственная отсталость, от умеренной до глубокой, задержка речи, расторможенность, особенности фенотипа: удлиненное лицо, макро- и долихоцефалия, гипоплазированная средняя часть лица, увеличенные щеки, толстые губы, нижняя часто вывернута. Большие, оттопыренные уши, большие кисти и стопы.
Умственная отсталость с ломкой Х- хромосомой, FRA-X синдром. Наиболее частая после болезни Дауна умственная отсталость, 1: 2000 -2500 новорожденных, мальчиков в 2 -3 раза больше, болеют тяжелее. Признаки: Умственная отсталость, от умеренной до глубокой, задержка речи, расторможенность, особенности фенотипа: удлиненное лицо, макро- и долихоцефалия, гипоплазированная средняя часть лица, увеличенные щеки, толстые губы, нижняя часто вывернута. Большие, оттопыренные уши, большие кисти и стопы.
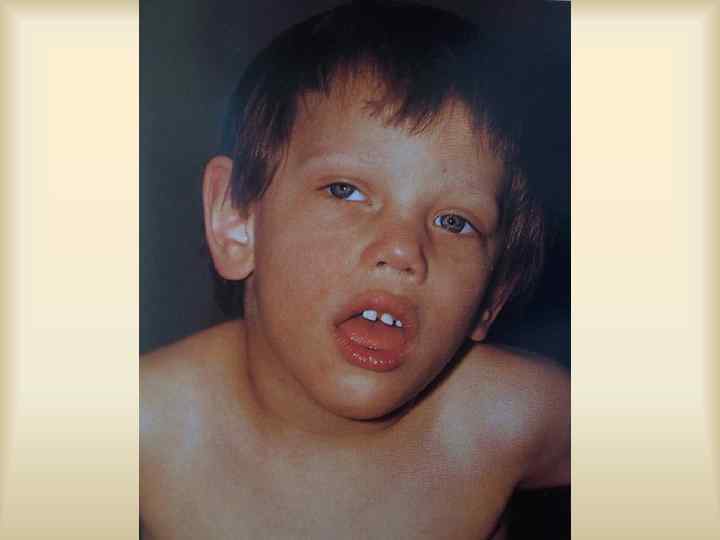
 Макроорхидизм, снижение половой активности. Признаки врожденной дисплазии соединительной ткани – плоскостопие, гипермобильность кожи, ПМК, слабость связочного аппарата суставов(вывихи и подвывихи), кривошея, сколиоз, кифоз, мышечная гипотония. Большинство социально адаптированы, выполняют несложную работу.
Макроорхидизм, снижение половой активности. Признаки врожденной дисплазии соединительной ткани – плоскостопие, гипермобильность кожи, ПМК, слабость связочного аппарата суставов(вывихи и подвывихи), кривошея, сколиоз, кифоз, мышечная гипотония. Большинство социально адаптированы, выполняют несложную работу.
 Цитогенетически – ломкость Хq в виде спутника, выявляемая при культивировании лимфоцитов в условиях дефицита фолиевой кислоты. Считалось, что заболевание Х-сц. рецессивное, но болели и женщины, отмечалась антиципация в родословной. Обнаружена экспансия тринуклеотидных повторов CGG в 5 нетранслируемой области гена FMR – 1 (fragile mental retardation). При количестве повторов более 200 не включается синтез белка, контролирующего функцию клеток ЦНС.
Цитогенетически – ломкость Хq в виде спутника, выявляемая при культивировании лимфоцитов в условиях дефицита фолиевой кислоты. Считалось, что заболевание Х-сц. рецессивное, но болели и женщины, отмечалась антиципация в родословной. Обнаружена экспансия тринуклеотидных повторов CGG в 5 нетранслируемой области гена FMR – 1 (fragile mental retardation). При количестве повторов более 200 не включается синтез белка, контролирующего функцию клеток ЦНС.
 Относится к группе наследственных нарушений биосинтеза стероидных гормонов. Известно несколько разновидностей наследственных дефицитов ферментов, обеспечивающих синтез стероидов (21 гидроксилаза, 11 -гидроксилаза, 17 -гидроксилаза и др. ). Все варианты этой наследственной патологии наследуются по аутосомнорецессивному типу. Ген стероид-21 гидроксилазы картирован (6 р21. 3).
Относится к группе наследственных нарушений биосинтеза стероидных гормонов. Известно несколько разновидностей наследственных дефицитов ферментов, обеспечивающих синтез стероидов (21 гидроксилаза, 11 -гидроксилаза, 17 -гидроксилаза и др. ). Все варианты этой наследственной патологии наследуются по аутосомнорецессивному типу. Ген стероид-21 гидроксилазы картирован (6 р21. 3).
 1: 5000") Частота дефицита 21 -гидроксилазы (определяет до 90% всех случаев врожденной гиперплазии надпочечников) 1: 5000 новорожденных. Известны два классических варианта этой болезни - сольтеряющая и простая вирильная форма. Другие формы встречаются значительно реже.
Частота дефицита 21 -гидроксилазы (определяет до 90% всех случаев врожденной гиперплазии надпочечников) 1: 5000 новорожденных. Известны два классических варианта этой болезни - сольтеряющая и простая вирильная форма. Другие формы встречаются значительно реже.
.") Сольтеряющая форма характеризуется полным дефицитом фермента и проявляется в нарушении солевого обмена (дефицит минералокортикоидов). В патологический процесс вовлекается ренин-альдостероновая система. У новорожденного отмечаются срыгивание, рвота, симптомы недостаточности периферического кровообращения, сонливость, потеря массы тела. Обезвоживание вызывает повышенную жажду. Биохимические исследование выявляет гиперкалиемию, гипонатриемию, ацидоз.
Сольтеряющая форма характеризуется полным дефицитом фермента и проявляется в нарушении солевого обмена (дефицит минералокортикоидов). В патологический процесс вовлекается ренин-альдостероновая система. У новорожденного отмечаются срыгивание, рвота, симптомы недостаточности периферического кровообращения, сонливость, потеря массы тела. Обезвоживание вызывает повышенную жажду. Биохимические исследование выявляет гиперкалиемию, гипонатриемию, ацидоз.
 Простая вирильная форма характеризуется прогрессирующей вирилизацией, ускоренным соматическим развитием, повышенной экскрецией гормонов коры надпочечников. У новорожденных девочек при кариотипе 46, ХХ отмечается различная степень маскулинизации (от умеренной гипертрофии клитора до полного срастания губно-мошоночных складок с формированием мошонки и пениса). У мальчиков заболевание проявляется лишь на 5 -7 -м году жизни преждевременным половым развитием.
Простая вирильная форма характеризуется прогрессирующей вирилизацией, ускоренным соматическим развитием, повышенной экскрецией гормонов коры надпочечников. У новорожденных девочек при кариотипе 46, ХХ отмечается различная степень маскулинизации (от умеренной гипертрофии клитора до полного срастания губно-мошоночных складок с формированием мошонки и пениса). У мальчиков заболевание проявляется лишь на 5 -7 -м году жизни преждевременным половым развитием.
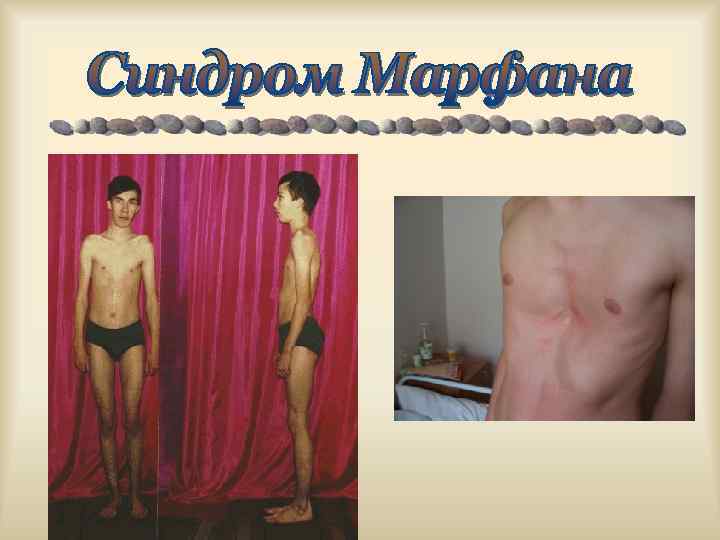
. В одном гене") Аутосомно-доминантная болезнь, причиной которой является мутация гена фибриллина (15 q 21). В одном гене описано более 10 вариантов мутаций, что обусловливает крайнюю вариабельность синдрома. Семейные случаи составляют 75% всех Популяционная частота 1: 10 000. наблюдений.
Аутосомно-доминантная болезнь, причиной которой является мутация гена фибриллина (15 q 21). В одном гене описано более 10 вариантов мутаций, что обусловливает крайнюю вариабельность синдрома. Семейные случаи составляют 75% всех Популяционная частота 1: 10 000. наблюдений.
 При СМ нарушается синтеза белка фибриллина и соединительная ткань обладает повышенной растяжимостью. тика разнообразна. Наиболее специфическими для диагностики СМ являются нарушения в скелете, вывих хрусталика, пороки развития сердечнососудистой системы, эктазия твердой мозговой оболочки.
При СМ нарушается синтеза белка фибриллина и соединительная ткань обладает повышенной растяжимостью. тика разнообразна. Наиболее специфическими для диагностики СМ являются нарушения в скелете, вывих хрусталика, пороки развития сердечнососудистой системы, эктазия твердой мозговой оболочки.
 Основные нарушения скелетно-мышечной системы: арахнодактилия, высокий рост, длинные конечности, деформация позвоночника (сколиоз, грудной лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, "куриная" грудь), ненормальная подвижность суставов, плоская стопа, высокое арковидное небо, недоразвитие впадины, мышечная гипотония. вертлужной
Основные нарушения скелетно-мышечной системы: арахнодактилия, высокий рост, длинные конечности, деформация позвоночника (сколиоз, грудной лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, "куриная" грудь), ненормальная подвижность суставов, плоская стопа, высокое арковидное небо, недоразвитие впадины, мышечная гипотония. вертлужной
 Основные симптомы поражения глазной системы: вывих хрусталика, миопия, отслоение сетчатки, длина уплощение оси глазного роговицы, яблока. увеличенная В сердечно- сосудистой системе обнаруживают: аортальную регургитацию, аневризму восходящей части аорты, расслоение аорты, пролапс митрального клапана, кальцификацию митрального отверстия, дизритмию.
Основные симптомы поражения глазной системы: вывих хрусталика, миопия, отслоение сетчатки, длина уплощение оси глазного роговицы, яблока. увеличенная В сердечно- сосудистой системе обнаруживают: аортальную регургитацию, аневризму восходящей части аорты, расслоение аорты, пролапс митрального клапана, кальцификацию митрального отверстия, дизритмию.
 Для заболевания характерны: паховые грыжи, спонтанный пневмоторакс, аномалии развития нервной системы. Диагностические критерии для диагноза СМ должны строго соблюдаться, так как ряд других врожденных дисплазий соединительной ткани могут быть приняты за СМ. Помогает в постановке диагноза наличие этого заболевания у родственника 1 -й степени родства.
Для заболевания характерны: паховые грыжи, спонтанный пневмоторакс, аномалии развития нервной системы. Диагностические критерии для диагноза СМ должны строго соблюдаться, так как ряд других врожденных дисплазий соединительной ткани могут быть приняты за СМ. Помогает в постановке диагноза наличие этого заболевания у родственника 1 -й степени родства.

 - синдром дисплазии соединительной Аутосомно-доминантное ткани. наследование, при аутосомно-рецессивном типе наследования") Синдром Элерса-Данлоса (СЭД) - синдром дисплазии соединительной Аутосомно-доминантное ткани. наследование, при аутосомно-рецессивном типе наследования имеет место лизилгидроксилазы, хромосомой лизилоксидазы. типе недостаточность при СЭД сцепленном с Х- недостаточность
Синдром Элерса-Данлоса (СЭД) - синдром дисплазии соединительной Аутосомно-доминантное ткани. наследование, при аутосомно-рецессивном типе наследования имеет место лизилгидроксилазы, хромосомой лизилоксидазы. типе недостаточность при СЭД сцепленном с Х- недостаточность
 По клинике различают пять форм заболевания, четыре из которых имеют АД наследование. Проявляется заболевание гиперэластичностью и ранимостью кожи, ее гиперпигментацией, гипермобильностью суставов, сколиозом, травматизацией кожи. Часто наблюдается пролапс митрального клапана, генерализованный периодонит, геморрагический синдром. Возможно нарушение интеллекта. Первая и вторая формы характеризуются развернутой симптоматикой заболевания, при третьей - преобладает гипермобильность суставов, при 4 -й рецидивирующие гематомы, 5 -й - гиперэластическая кожа.
По клинике различают пять форм заболевания, четыре из которых имеют АД наследование. Проявляется заболевание гиперэластичностью и ранимостью кожи, ее гиперпигментацией, гипермобильностью суставов, сколиозом, травматизацией кожи. Часто наблюдается пролапс митрального клапана, генерализованный периодонит, геморрагический синдром. Возможно нарушение интеллекта. Первая и вторая формы характеризуются развернутой симптоматикой заболевания, при третьей - преобладает гипермобильность суставов, при 4 -й рецидивирующие гематомы, 5 -й - гиперэластическая кожа.

 Обусловлена мутацией гена рецептора фактора роста фибробластов, вызывающей отклонения в активности некоторых ферментов (5 -нуклеотидазы, глюкозо-6 -фосфатазы), в результате чего нарушается рост и развитие хрящевой ткани в эпифизах трубчатых костей и в основании черепа. Тип наследования - аутосомнодоминантный. Популяционная частота - 1: 100 000. Не менее 80% случаев болезни обусловлено новой мутацией гена EGFR 3, локализованного на 4 р.
Обусловлена мутацией гена рецептора фактора роста фибробластов, вызывающей отклонения в активности некоторых ферментов (5 -нуклеотидазы, глюкозо-6 -фосфатазы), в результате чего нарушается рост и развитие хрящевой ткани в эпифизах трубчатых костей и в основании черепа. Тип наследования - аутосомнодоминантный. Популяционная частота - 1: 100 000. Не менее 80% случаев болезни обусловлено новой мутацией гена EGFR 3, локализованного на 4 р.
 при сохранении большой") Характерными признаками заболевания являются низкий рост (120 -130 см у взрослых) при сохранении большой нормальной череп с длины выступающим туловища, затылком, запавшая переносица. Конечности укорочены, в основном за счет проксимальных отделов бедренной и плечевой костей, кисти широкие и короткие. Дети отстают в моторном развитии, интеллект, как правило, не страдает.
Характерными признаками заболевания являются низкий рост (120 -130 см у взрослых) при сохранении большой нормальной череп с длины выступающим туловища, затылком, запавшая переносица. Конечности укорочены, в основном за счет проксимальных отделов бедренной и плечевой костей, кисти широкие и короткие. Дети отстают в моторном развитии, интеллект, как правило, не страдает.
 Тяжелое заболевание, проявляющееся мышечной слабостью и повышенным содержанием в плазме крови креатинфосфокиназы (в 10 -100 раз). Встречается с частотой 1: 3 500 новорожденных мальчиков. рецессивный. Ген МД картирован в области Хр21 и детально изучен, что позволяет проводить молекулярно-генетическую диагностику.
Тяжелое заболевание, проявляющееся мышечной слабостью и повышенным содержанием в плазме крови креатинфосфокиназы (в 10 -100 раз). Встречается с частотой 1: 3 500 новорожденных мальчиков. рецессивный. Ген МД картирован в области Хр21 и детально изучен, что позволяет проводить молекулярно-генетическую диагностику.
 Для МД характерно раннее, в возрасте 35 -и лет, начало заболевания: нарастающая слабость в постепенным икроножные плечевого Появляется мышцах бедер переходом мышцы, пояса, утиная и процесса мышцы спины, таза на верхнего живота походка. с и др. Заболевание неуклонно прогрессирует, дети оказываются прикованными к постели с 10 -11 -летнего
Для МД характерно раннее, в возрасте 35 -и лет, начало заболевания: нарастающая слабость в постепенным икроножные плечевого Появляется мышцах бедер переходом мышцы, пояса, утиная и процесса мышцы спины, таза на верхнего живота походка. с и др. Заболевание неуклонно прогрессирует, дети оказываются прикованными к постели с 10 -11 -летнего
 Наблюдается псевдогипертрофия икроножных и ягодичных мышц за счет замещения мышечной ткани соединительной и жировой. Во многих случаях развивается сгибательная мышечная контрактура бедренных и коленных суставов и суставов верхних конечностей вследствие атрофии мышц. Рано снижаются глубокие сухожильные рефлексы. Имеется тенденция к некоторому снижению умственных способностей. Продолжительность жизни больных - 20 -35 лет.
Наблюдается псевдогипертрофия икроножных и ягодичных мышц за счет замещения мышечной ткани соединительной и жировой. Во многих случаях развивается сгибательная мышечная контрактура бедренных и коленных суставов и суставов верхних конечностей вследствие атрофии мышц. Рано снижаются глубокие сухожильные рефлексы. Имеется тенденция к некоторому снижению умственных способностей. Продолжительность жизни больных - 20 -35 лет.