

adrenal gland disorders.ppt
- Количество слайдов: 78
 Disorders of adrenal gland in children and teenagers. Pediatry department Assistant Golovko Tatyana
Disorders of adrenal gland in children and teenagers. Pediatry department Assistant Golovko Tatyana
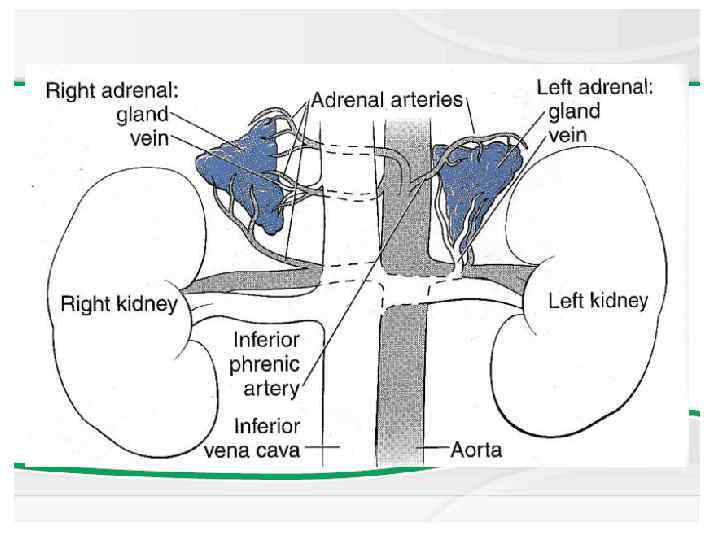
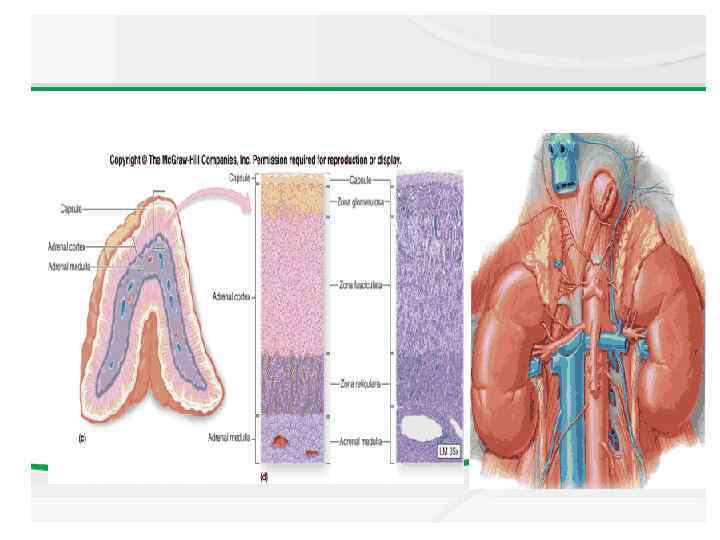
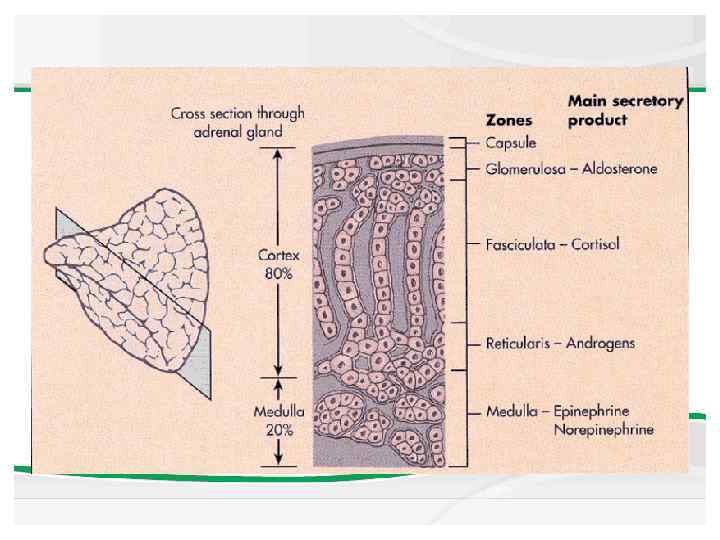
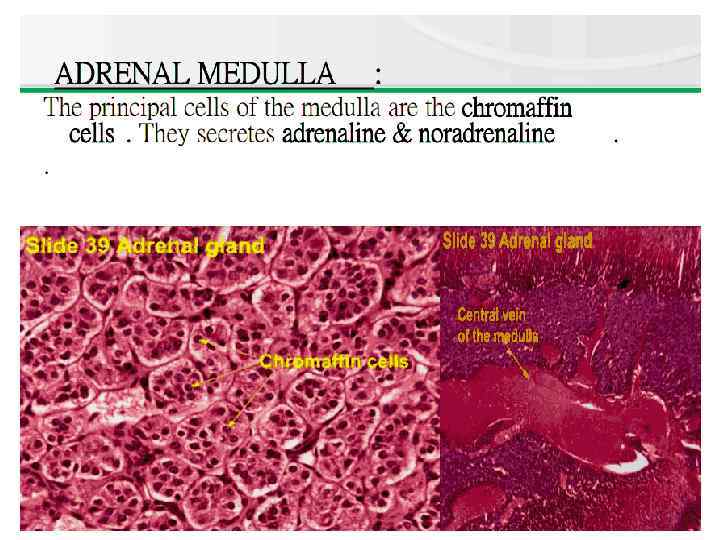
 Anatomy The adrenal cortex is divided into 3 major anatomic zones. The zone glomerulosa produces aldosterone, and the zone fasciculata and reticularis together produce cortisol and adrenal androgens. A fetal zone produces dehydroepiandrosterone (DHEA), a precursor of both androgens and estrogens. This zone involutes within the first few months of postnatal life.
Anatomy The adrenal cortex is divided into 3 major anatomic zones. The zone glomerulosa produces aldosterone, and the zone fasciculata and reticularis together produce cortisol and adrenal androgens. A fetal zone produces dehydroepiandrosterone (DHEA), a precursor of both androgens and estrogens. This zone involutes within the first few months of postnatal life.


 Addison’s Disease • 1 st described in 1855 by Dr. Thomas Addison; • Refers to acquired primary adrenal insufficiency; • Does not have specific etiology (Usually autoimmune (~80%). Addison’s Normal
Addison’s Disease • 1 st described in 1855 by Dr. Thomas Addison; • Refers to acquired primary adrenal insufficiency; • Does not have specific etiology (Usually autoimmune (~80%). Addison’s Normal
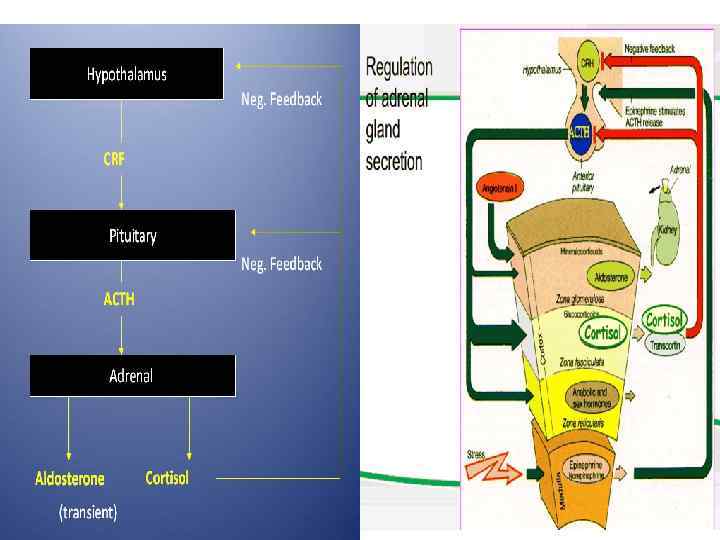
 - primary, which occurs when the adrenal gland itself is") Adrenal insufficiency (Addison disease) - primary, which occurs when the adrenal gland itself is dysfunctional, - secondary, also called central adrenal insufficiency, which occurs when a lack of secretion of corticotropin-releasing hormone (CRH) from the hypothalamus or of adrenocorticotropic hormone (ACTH) from the pituitary leads to hypofunction of the adrenal cortex.
Adrenal insufficiency (Addison disease) - primary, which occurs when the adrenal gland itself is dysfunctional, - secondary, also called central adrenal insufficiency, which occurs when a lack of secretion of corticotropin-releasing hormone (CRH) from the hypothalamus or of adrenocorticotropic hormone (ACTH) from the pituitary leads to hypofunction of the adrenal cortex.
 • Congenital; • Acquired. Most cases of adrenal insufficiency (Addison") Adrenal insufficiency (Addison disease) • Congenital; • Acquired. Most cases of adrenal insufficiency (Addison disease) are iatrogenic, caused by long-term administration of glucocorticoids. A 2 weeks' exposure to pharmacologic doses of glucocorticoids can suppress the corticotropinreleasing hormone (CRH)–adrenocorticotropic hormone (ACTH)–adrenal axis.
Adrenal insufficiency (Addison disease) • Congenital; • Acquired. Most cases of adrenal insufficiency (Addison disease) are iatrogenic, caused by long-term administration of glucocorticoids. A 2 weeks' exposure to pharmacologic doses of glucocorticoids can suppress the corticotropinreleasing hormone (CRH)–adrenocorticotropic hormone (ACTH)–adrenal axis.
 Acquired primary adrenal insufficiency In developed countries, the most common cause of adrenal insufficiency (Addison disease) is autoimmune destruction of the adrenal cortex. This disorder may occur in isolation or may be part of a polyglandular autoimmune disorder (PGAD). Patients with type 1 PGAD usually present in the first decade of life with mucocutaneous candidiasis or hypoparathyroidism. This is an autosomal recessive disorder that involves the AIRE gene on chromosome 21 and presents with all or some of the following features:
Acquired primary adrenal insufficiency In developed countries, the most common cause of adrenal insufficiency (Addison disease) is autoimmune destruction of the adrenal cortex. This disorder may occur in isolation or may be part of a polyglandular autoimmune disorder (PGAD). Patients with type 1 PGAD usually present in the first decade of life with mucocutaneous candidiasis or hypoparathyroidism. This is an autosomal recessive disorder that involves the AIRE gene on chromosome 21 and presents with all or some of the following features:
 • • • Chronic mucocutaneous candidiasis; Hypoparathyroidism; Adrenal failure; Gonadal failure; Vitiligo; Alopecia; Hypothyroidism; Type 1 diabetes mellitus; Pernicious anemia; Steatorrhea.
• • • Chronic mucocutaneous candidiasis; Hypoparathyroidism; Adrenal failure; Gonadal failure; Vitiligo; Alopecia; Hypothyroidism; Type 1 diabetes mellitus; Pernicious anemia; Steatorrhea.
 consists of type 1 diabetes mellitus, autoimmune thyroid disease,") Type 2 PGAD (Schmidt syndrome) consists of type 1 diabetes mellitus, autoimmune thyroid disease, and adrenal failure. Individuals with this condition generally present in the second or third decades of life, although some components of the syndrome may be present in the pediatric age group. Type 2 PGAD is transmitted as an autosomal disorder with variable penetrance. Addison disease should be considered in patients with type 1 diabetes and unexplained fatigue, hypotension, hypoglycemia, hyponatremia and hyperkalemia.
Type 2 PGAD (Schmidt syndrome) consists of type 1 diabetes mellitus, autoimmune thyroid disease, and adrenal failure. Individuals with this condition generally present in the second or third decades of life, although some components of the syndrome may be present in the pediatric age group. Type 2 PGAD is transmitted as an autosomal disorder with variable penetrance. Addison disease should be considered in patients with type 1 diabetes and unexplained fatigue, hypotension, hypoglycemia, hyponatremia and hyperkalemia.
 Other acquired causes of adrenal failure include the following: • Adrenal hemorrhage; • Infections (eg, tuberculosis [TB], human immunodeficiency virus [HIV] infection); • Neoplastic destruction; • Metabolic disorders (eg, various forms of adrenal leukodystrophy , Wolman disease, Smith-Lemli-Opitz syndrome); • Administration of the anesthetic agent etomidate.
Other acquired causes of adrenal failure include the following: • Adrenal hemorrhage; • Infections (eg, tuberculosis [TB], human immunodeficiency virus [HIV] infection); • Neoplastic destruction; • Metabolic disorders (eg, various forms of adrenal leukodystrophy , Wolman disease, Smith-Lemli-Opitz syndrome); • Administration of the anesthetic agent etomidate.
 Congenital primary adrenal insufficiency Congenital Addison disease may occur as a result of adrenal hypoplasia or hyperplasia. Inherited as an X-linked disorder, adrenal hypoplasia congenita is caused by deletion or mutation of the DAX 1/NR 0 B 1 gene on chromosome Xp 21. 2, and is additionally associated with hypogonadotrophic hypogonadism and primary defects in sperm production. Congenital adrenal hyperplasia results from a deficiency of one of several enzymes required for adrenal synthesis of cortisol. Symptoms of adrenal insufficiency (Addison disease) most often develop with combined deficiencies of cortisol and aldosterone. The most prevalent form of congenital adrenal hyperplasia is caused by a deficiency in steroid 21 -hydroxylase.
Congenital primary adrenal insufficiency Congenital Addison disease may occur as a result of adrenal hypoplasia or hyperplasia. Inherited as an X-linked disorder, adrenal hypoplasia congenita is caused by deletion or mutation of the DAX 1/NR 0 B 1 gene on chromosome Xp 21. 2, and is additionally associated with hypogonadotrophic hypogonadism and primary defects in sperm production. Congenital adrenal hyperplasia results from a deficiency of one of several enzymes required for adrenal synthesis of cortisol. Symptoms of adrenal insufficiency (Addison disease) most often develop with combined deficiencies of cortisol and aldosterone. The most prevalent form of congenital adrenal hyperplasia is caused by a deficiency in steroid 21 -hydroxylase.
 caused by") Lipoid adrenal hyperplasia is another rare form of adrenal insufficiency (Addison disease) caused by a mutation in the steroid acute regulatory protein (ie, STAR protein) or a mutation in the cholesterol side-chain cleavage gene (at the cytochrome P 450 [CYP] 11 A locus). This disease causes a defective synthesis of all adrenocortical hormones. In its complete form, the disease is lethal.
Lipoid adrenal hyperplasia is another rare form of adrenal insufficiency (Addison disease) caused by a mutation in the steroid acute regulatory protein (ie, STAR protein) or a mutation in the cholesterol side-chain cleavage gene (at the cytochrome P 450 [CYP] 11 A locus). This disease causes a defective synthesis of all adrenocortical hormones. In its complete form, the disease is lethal.
 is rare in children. It may result from:") Primary adrenal cortical insufficiency (Addison disease) is rare in children. It may result from: • An autoimmune process, sometimes in association with other autoimmune endocrine disorders, e. g. diabetes mellitus, hypothyroidism, hypoparathyroidism • Haemorrhage/infarction – neonatal, meningococcal septicaemia (usually fatal) • X-linked adrenoleucodystrophy, a rare neurodegenerative metabolic disorder • Tuberculosis, now rare. Adrenal insufficiency may also be secondary to hypopituitarism from hypothalamic–pituitary disease or from hypothalamic–pituitary–adrenal suppression following longterm corticosteroid therapy.
Primary adrenal cortical insufficiency (Addison disease) is rare in children. It may result from: • An autoimmune process, sometimes in association with other autoimmune endocrine disorders, e. g. diabetes mellitus, hypothyroidism, hypoparathyroidism • Haemorrhage/infarction – neonatal, meningococcal septicaemia (usually fatal) • X-linked adrenoleucodystrophy, a rare neurodegenerative metabolic disorder • Tuberculosis, now rare. Adrenal insufficiency may also be secondary to hypopituitarism from hypothalamic–pituitary disease or from hypothalamic–pituitary–adrenal suppression following longterm corticosteroid therapy.
 can live") Prognosis With proper treatment and compliance, patients with adrenal insufficiency (Addison disease) can live a normal life span without limitations. However, the prognosis for an untreated patient with adrenal insufficiency (Addison disease) is poor. Some studies have found that those with very high concentrations of cortisol have a worse prognosis and a higher complication rate of secondary sepsis or intestinal perforation. Death is a common outcome—usually from hypotension or cardiac arrhythmia secondary to hyperkalemia—unless replacement steroid therapy is begun.
Prognosis With proper treatment and compliance, patients with adrenal insufficiency (Addison disease) can live a normal life span without limitations. However, the prognosis for an untreated patient with adrenal insufficiency (Addison disease) is poor. Some studies have found that those with very high concentrations of cortisol have a worse prognosis and a higher complication rate of secondary sepsis or intestinal perforation. Death is a common outcome—usually from hypotension or cardiac arrhythmia secondary to hyperkalemia—unless replacement steroid therapy is begun.
 generally present with acute") Acute adrenal insufficiency • Patients with adrenal insufficiency (Addison disease) generally present with acute dehydration, hypotension (especially orthostatic hypotension and tachycardia), symptomatic hypoglycemia, or altered mental status. These signs may occur in conjunction with acute sepsis or disseminated intravascular coagulation or in a patient after a traumatic delivery. • In infants, acute adrenal insufficiency (Addison disease) may occur in the context of serious illness (eg, sepsis), prolonged and difficult labor, or traumatic delivery. Tuberculosis (TB), meningococcemia, or any severe septicemia may also result in adrenal insufficiency (Addison disease). However, adrenal insufficiency (Addison disease) may occur without concomitant illness when it is due to congenital adrenal hyperplasia or congenital adrenal hypoplasia.
Acute adrenal insufficiency • Patients with adrenal insufficiency (Addison disease) generally present with acute dehydration, hypotension (especially orthostatic hypotension and tachycardia), symptomatic hypoglycemia, or altered mental status. These signs may occur in conjunction with acute sepsis or disseminated intravascular coagulation or in a patient after a traumatic delivery. • In infants, acute adrenal insufficiency (Addison disease) may occur in the context of serious illness (eg, sepsis), prolonged and difficult labor, or traumatic delivery. Tuberculosis (TB), meningococcemia, or any severe septicemia may also result in adrenal insufficiency (Addison disease). However, adrenal insufficiency (Addison disease) may occur without concomitant illness when it is due to congenital adrenal hyperplasia or congenital adrenal hypoplasia.
 usually have chronic") Chronic adrenal insufficiency • Patients with chronic adrenal insufficiency (Addison disease) usually have chronic fatigue, anorexia, asthenia, nausea, vomiting, loss of appetite, weight loss, recurring abdominal pain, and weakness and a lack of energy. Increased skin pigmentation and salt craving are common among individuals with chronic primary adrenal insufficiency. Salt craving is a symptom typical of patients with dysfunction of the zona glomerulosa; this craving may be the first sign of autoimmune adrenal destruction. • Signs of weight loss may be evident. If the patient is not frankly hypotensive, he or she may have orthostatic hypotension. • Some patients lose pubic and axillary hair because adrenal androgens support growth of body hair in these areas.
Chronic adrenal insufficiency • Patients with chronic adrenal insufficiency (Addison disease) usually have chronic fatigue, anorexia, asthenia, nausea, vomiting, loss of appetite, weight loss, recurring abdominal pain, and weakness and a lack of energy. Increased skin pigmentation and salt craving are common among individuals with chronic primary adrenal insufficiency. Salt craving is a symptom typical of patients with dysfunction of the zona glomerulosa; this craving may be the first sign of autoimmune adrenal destruction. • Signs of weight loss may be evident. If the patient is not frankly hypotensive, he or she may have orthostatic hypotension. • Some patients lose pubic and axillary hair because adrenal androgens support growth of body hair in these areas.
 activity from adrenocorticotropic hormone (ACTH) causes the hyperpigmentation. Hyperpigmentation is") Excess melanocyte-stimulating hormone (MSH) activity from adrenocorticotropic hormone (ACTH) causes the hyperpigmentation. Hyperpigmentation is noted in patients with secondary or central adrenal insufficiency (Addison disease) due to ACTH or corticotropinreleasing hormone (CRH) deficiency, because these conditions do not elevate serum ACTH concentrations. Left photograph shows hyperpigmentation on the dorsum of a patient's hand before the treatment of primary adrenal insufficiency. Right photograph shows normal pigmentation after treatment. Left photograph shows a patient with Addison disease who has prominent pigmentation in areas not exposed to the sun, such as the palmar creases. Right photograph shows normal pigmentation after treatment.
Excess melanocyte-stimulating hormone (MSH) activity from adrenocorticotropic hormone (ACTH) causes the hyperpigmentation. Hyperpigmentation is noted in patients with secondary or central adrenal insufficiency (Addison disease) due to ACTH or corticotropinreleasing hormone (CRH) deficiency, because these conditions do not elevate serum ACTH concentrations. Left photograph shows hyperpigmentation on the dorsum of a patient's hand before the treatment of primary adrenal insufficiency. Right photograph shows normal pigmentation after treatment. Left photograph shows a patient with Addison disease who has prominent pigmentation in areas not exposed to the sun, such as the palmar creases. Right photograph shows normal pigmentation after treatment.
 Autoimmune adrenal insufficiency, adrenal insufficiency from adrenoleukodystrophy • autoimmune adrenal insufficiency or adrenal insufficiency due to adrenoleukodystrophy, chronic infections (eg, human immunodeficiency virus (HIV) infection, TB, fungal infection), or infiltrative lesions usually present with chronic symptoms (eg, fatigue, anorexia, abdominal pain). However, an acute adrenal crisis may exacerbate the symptoms.
Autoimmune adrenal insufficiency, adrenal insufficiency from adrenoleukodystrophy • autoimmune adrenal insufficiency or adrenal insufficiency due to adrenoleukodystrophy, chronic infections (eg, human immunodeficiency virus (HIV) infection, TB, fungal infection), or infiltrative lesions usually present with chronic symptoms (eg, fatigue, anorexia, abdominal pain). However, an acute adrenal crisis may exacerbate the symptoms.
 Differential Diagnoses • • 3 -Beta-Hydroxysteroid Dehydrogenase Deficiency; Adrenal Hypoplasia; Birth Trauma; Chronic Fatigue Syndrome; Congenital Adrenal Hyperplasia; Familial Glucocorticoid Deficiency; Hypopituitarism; Pseudohypoaldosteronism.
Differential Diagnoses • • 3 -Beta-Hydroxysteroid Dehydrogenase Deficiency; Adrenal Hypoplasia; Birth Trauma; Chronic Fatigue Syndrome; Congenital Adrenal Hyperplasia; Familial Glucocorticoid Deficiency; Hypopituitarism; Pseudohypoaldosteronism.
 Primary adrenal insufficiency: Laboratory findings • • • Hyponatremia; Hyperkalemia; Hypoglycemia; Narrow cardiac silhouette on Chest-XR; Low voltage EKG.
Primary adrenal insufficiency: Laboratory findings • • • Hyponatremia; Hyperkalemia; Hypoglycemia; Narrow cardiac silhouette on Chest-XR; Low voltage EKG.
 is confirmed if the") Criteria for diagnosis A diagnosis of adrenal insufficiency (Addison disease) is confirmed if the serum cortisol level is less than 18 mcg/d. L in the presence of a markedly elevated serum adrenocorticotropic hormone (ACTH) concentration and plasma renin activity. Based on normative data of children of various ages, adrenal insufficiency (Addison disease) is likely if the serum cortisol concentration is less than 18 mcg/d. L 30 -60 minutes after administration of 250 mcg of cosyntropin (synthetic ACTH).
Criteria for diagnosis A diagnosis of adrenal insufficiency (Addison disease) is confirmed if the serum cortisol level is less than 18 mcg/d. L in the presence of a markedly elevated serum adrenocorticotropic hormone (ACTH) concentration and plasma renin activity. Based on normative data of children of various ages, adrenal insufficiency (Addison disease) is likely if the serum cortisol concentration is less than 18 mcg/d. L 30 -60 minutes after administration of 250 mcg of cosyntropin (synthetic ACTH).
 stimulation test The standard ovine corticotropin-releasing hormone (CRH) stimulation test (1") Corticotropin-releasing hormone (CRH) stimulation test The standard ovine corticotropin-releasing hormone (CRH) stimulation test (1 mcg/kg over 1 min) may be helpful in the differential diagnosis of adrenal insufficiency (Addison disease). A lack of a 2 -fold increase in serum adrenocorticotropic hormone (ACTH) concentration indicates pituitary dysfunction. A 2 -fold or greater rise in ACTH without a concomitant rise in serum cortisol to more than 18 -20 mcg/d. L implies primary adrenal insufficiency (Addison disease).
Corticotropin-releasing hormone (CRH) stimulation test The standard ovine corticotropin-releasing hormone (CRH) stimulation test (1 mcg/kg over 1 min) may be helpful in the differential diagnosis of adrenal insufficiency (Addison disease). A lack of a 2 -fold increase in serum adrenocorticotropic hormone (ACTH) concentration indicates pituitary dysfunction. A 2 -fold or greater rise in ACTH without a concomitant rise in serum cortisol to more than 18 -20 mcg/d. L implies primary adrenal insufficiency (Addison disease).
 Insulin-tolerance test An insulin-tolerance test requires an intravenous administration of insulin (usually regular insulin 0. 05 -0. 15 U/kg) to induce a 50% reduction in blood sugar concentration. Cortisol and glucose concentrations are measured every 15 minutes for 60 minutes. The test is considered adequate if the blood sugars level decreases by at least 50%. In response to the hypoglycemic stimulus, serum or plasma cortisol concentrations should rise to more than 18 -20 mcg/d. L.
Insulin-tolerance test An insulin-tolerance test requires an intravenous administration of insulin (usually regular insulin 0. 05 -0. 15 U/kg) to induce a 50% reduction in blood sugar concentration. Cortisol and glucose concentrations are measured every 15 minutes for 60 minutes. The test is considered adequate if the blood sugars level decreases by at least 50%. In response to the hypoglycemic stimulus, serum or plasma cortisol concentrations should rise to more than 18 -20 mcg/d. L.
 Metyrapone stimulation Standard metyrapone stimulation tests involve administering metyrapone 300 mg/m 2 in 6 divided doses over 24 hours. Because metyrapone inhibits 11 hydroxylase, which is involved in the last enzymatic step in cortisol synthesis, plasma levels of the cortisol precursor, 11 -deoxycortisol, increase. A normal response is a rise in 11 -deoxycortisol concentrations to more than 10. 5 mcg/d. L 4 hours after the last dose of metyrapone is given or a 2 -fold to 3 -fold increase in 24 -hour urinary concentrations of 17 hydroxycorticosteroid (which include tetrahydro compound S, a urinary metabolite of 11 -deoxycortisol) on the day of or the day after the administration of metyrapone.
Metyrapone stimulation Standard metyrapone stimulation tests involve administering metyrapone 300 mg/m 2 in 6 divided doses over 24 hours. Because metyrapone inhibits 11 hydroxylase, which is involved in the last enzymatic step in cortisol synthesis, plasma levels of the cortisol precursor, 11 -deoxycortisol, increase. A normal response is a rise in 11 -deoxycortisol concentrations to more than 10. 5 mcg/d. L 4 hours after the last dose of metyrapone is given or a 2 -fold to 3 -fold increase in 24 -hour urinary concentrations of 17 hydroxycorticosteroid (which include tetrahydro compound S, a urinary metabolite of 11 -deoxycortisol) on the day of or the day after the administration of metyrapone.
 is confirmed, antiadrenal antibodies, specifically") Antiadrenal Antibody Testing When primary adrenal insufficiency (Addison disease) is confirmed, antiadrenal antibodies, specifically anti-21 -hydroxylase antibodies, can confirm an autoimmune cause for the disorder. If results for antiadrenal antibodies are negative, search for another etiology, such as tuberculosis (TB), adrenal hemorrhage, or adrenoleukodystrophy.
Antiadrenal Antibody Testing When primary adrenal insufficiency (Addison disease) is confirmed, antiadrenal antibodies, specifically anti-21 -hydroxylase antibodies, can confirm an autoimmune cause for the disorder. If results for antiadrenal antibodies are negative, search for another etiology, such as tuberculosis (TB), adrenal hemorrhage, or adrenoleukodystrophy.
 scanning is the imaging study of choice in the evaluation of") Computed tomography (CT) scanning is the imaging study of choice in the evaluation of adrenal insufficiency (Addison disease) and helps to identify adrenal hemorrhage, calcifications (see the following image), or infiltrative disease. Abdominal radiography may reveal bilateral adrenal calcifications, which suggest a history of bilateral adrenal hemorrhage, tuberculosis (TB), or Wolman disease. Magnetic resonance imaging (MRI) is not as useful as CT scanning, and iodocholesterol scanning is also not particularly useful for adrenal insufficiency Ultrasonography is a poor imaging modality for investigating the adrenal glands.
Computed tomography (CT) scanning is the imaging study of choice in the evaluation of adrenal insufficiency (Addison disease) and helps to identify adrenal hemorrhage, calcifications (see the following image), or infiltrative disease. Abdominal radiography may reveal bilateral adrenal calcifications, which suggest a history of bilateral adrenal hemorrhage, tuberculosis (TB), or Wolman disease. Magnetic resonance imaging (MRI) is not as useful as CT scanning, and iodocholesterol scanning is also not particularly useful for adrenal insufficiency Ultrasonography is a poor imaging modality for investigating the adrenal glands.
. Mineralocorticoid") Treatment Glucocorticoid replacement is required in all forms of adrenal insufficiency (Addison disease). Mineralocorticoid replacement is required only in primary adrenal insufficiency, because aldosterone secretion is reduced in primary adrenal insufficiency but not in secondary (central) adrenal insufficiency. No surgical management is needed in most cases.
Treatment Glucocorticoid replacement is required in all forms of adrenal insufficiency (Addison disease). Mineralocorticoid replacement is required only in primary adrenal insufficiency, because aldosterone secretion is reduced in primary adrenal insufficiency but not in secondary (central) adrenal insufficiency. No surgical management is needed in most cases.
 Adrenal crisis Acute manifestation of Addison’s is called Addison Crisis. Adrenal crisis and severe acute adrenocortical insufficiency are often elusive diagnoses that may result in severe morbidity and mortality when undiagnosed or ineffectively treated.
Adrenal crisis Acute manifestation of Addison’s is called Addison Crisis. Adrenal crisis and severe acute adrenocortical insufficiency are often elusive diagnoses that may result in severe morbidity and mortality when undiagnosed or ineffectively treated.
 Causes of adrenal crisis • Prior steroid use. Rapid withdrawal of long-term steroid therapy Patients receiving doses close to normal physiologic levels require only 1 month to recover normal adrenal function; • Organisms associated with adrenal crisis: Haemophilus influenzae, Staphylococcus aureus, Streptococcus pneumonia, fungi); • Meningococcemia; • Severe physiologic stress (e. g. , sepsis, trauma, burns, surgery); • Azotemia; • Anticoagulants, hemorrhagic diathesis; • Newborn, complicated pregnancy; • Adrenocorticotropin therapy, known primary or secondary adrenocortical insufficiency; • AIDS; • Invasive or infiltrative disorders; • Tuberculosis; • Topical steroids: Risk of adrenal crisis occurs when used over a large surface area for a prolonged duration, using occlusive dressings and a highly potent drug; • Inhaled steroids.
Causes of adrenal crisis • Prior steroid use. Rapid withdrawal of long-term steroid therapy Patients receiving doses close to normal physiologic levels require only 1 month to recover normal adrenal function; • Organisms associated with adrenal crisis: Haemophilus influenzae, Staphylococcus aureus, Streptococcus pneumonia, fungi); • Meningococcemia; • Severe physiologic stress (e. g. , sepsis, trauma, burns, surgery); • Azotemia; • Anticoagulants, hemorrhagic diathesis; • Newborn, complicated pregnancy; • Adrenocorticotropin therapy, known primary or secondary adrenocortical insufficiency; • AIDS; • Invasive or infiltrative disorders; • Tuberculosis; • Topical steroids: Risk of adrenal crisis occurs when used over a large surface area for a prolonged duration, using occlusive dressings and a highly potent drug; • Inhaled steroids.
 Clinical presentation • Unexplained shock, usually refractory to fluid and pressor resuscitation; • Nausea, vomiting, diarrhea; • Hyperthermia or hypothermia; • Hypotension; • Sudden, severe abdominal pain, pain in back, belly or legs; • Loss of consciousness; • Can be fatal.
Clinical presentation • Unexplained shock, usually refractory to fluid and pressor resuscitation; • Nausea, vomiting, diarrhea; • Hyperthermia or hypothermia; • Hypotension; • Sudden, severe abdominal pain, pain in back, belly or legs; • Loss of consciousness; • Can be fatal.
; hyperkalemia, metabolic acidosis,") Laboratory Studies • Serum chemistry: hyponatremia is common (although not diagnostic); hyperkalemia, metabolic acidosis, and hypoglycemia also may be present. However, the absence of laboratory abnormalities does not exclude the diagnosis of adrenal crisis. • Serum cortisol: Less than 20 mcg/d. L in severe stress or after ACTH stimulation is indicative of adrenal insufficiency. • ACTH test (diagnostic): Determine baseline serum cortisol, then administer ACTH 250 mcg intravenous push (IVP), and then draw serum cortisol 30 and 60 minutes after ACTH administration. An increase of less than 9 mcg/d. L is considered diagnostic of adrenal insufficiency. • CBC: Anemia (mild and nonspecific), lymphocytosis, and eosinophilia (highly suggestive) may be present.
Laboratory Studies • Serum chemistry: hyponatremia is common (although not diagnostic); hyperkalemia, metabolic acidosis, and hypoglycemia also may be present. However, the absence of laboratory abnormalities does not exclude the diagnosis of adrenal crisis. • Serum cortisol: Less than 20 mcg/d. L in severe stress or after ACTH stimulation is indicative of adrenal insufficiency. • ACTH test (diagnostic): Determine baseline serum cortisol, then administer ACTH 250 mcg intravenous push (IVP), and then draw serum cortisol 30 and 60 minutes after ACTH administration. An increase of less than 9 mcg/d. L is considered diagnostic of adrenal insufficiency. • CBC: Anemia (mild and nonspecific), lymphocytosis, and eosinophilia (highly suggestive) may be present.
 Imaging Studies • Chest radiography: Assess for tuberculosis, histoplasmosis, malignant disease, sarcoid, and lymphoma. • Abdominal CT scanning: Visualize adrenal glands for hemorrhage (as in the image below), atrophy, infiltrative disorders, and metastatic disease. Adrenal hemorrhage appears as hyperdense, bilaterally enlarged adrenal glands. • Electrocardiography Prolongation of the QT interval can induce ventricular arrhythmias. Deep negative T waves have been described in acute adrenal crisis.
Imaging Studies • Chest radiography: Assess for tuberculosis, histoplasmosis, malignant disease, sarcoid, and lymphoma. • Abdominal CT scanning: Visualize adrenal glands for hemorrhage (as in the image below), atrophy, infiltrative disorders, and metastatic disease. Adrenal hemorrhage appears as hyperdense, bilaterally enlarged adrenal glands. • Electrocardiography Prolongation of the QT interval can induce ventricular arrhythmias. Deep negative T waves have been described in acute adrenal crisis.
 Medical Care Administration of glucocorticoids in supraphysiologic or stress doses is the only definitive therapy (hydrocortisone 10 -15 mg/kg/d or is an initial dose of 50 -75 mg/m 2 given intravenously, followed by 50 -75 mg/m 2/d divided in 4 intravenous doses. Hydrocortisone may be given intramuscularly if intravenous access is unavailable. However, intramuscular administration works slowly). Comparable stress doses of methylprednisolone are 10 -15 mg/m 2 and dexamethasone 1 -1. 5 mg/m 2 or prednisolone 2 -5 mg/kg/d IM or IV.
Medical Care Administration of glucocorticoids in supraphysiologic or stress doses is the only definitive therapy (hydrocortisone 10 -15 mg/kg/d or is an initial dose of 50 -75 mg/m 2 given intravenously, followed by 50 -75 mg/m 2/d divided in 4 intravenous doses. Hydrocortisone may be given intramuscularly if intravenous access is unavailable. However, intramuscular administration works slowly). Comparable stress doses of methylprednisolone are 10 -15 mg/m 2 and dexamethasone 1 -1. 5 mg/m 2 or prednisolone 2 -5 mg/kg/d IM or IV.
, because") Dexamethasone is preferable for patients with suspected but unproved adrenal insufficiency (Addison disease), because the physician can simultaneously treat the patient while performing a diagnostic cosyntropin stimulation test. Methylprednisolone and dexamethasone have negligible mineralocorticoid effects. Large doses of hydrocortisone (ie, even double or triple the stress doses previously mentioned) are preferred if the patient is hypovolemic, hyponatremic, or hyperkalemic, due to the mineralocorticoid effects of hydrocortisone (lacking in prednisone or dexamethasone). No parenteral form of a mineralocorticoid is currently available in the United States. However, if the patient has good gastrointestinal function, fludrocortisone 0. 1 -0. 2 mg may be orally administered.
Dexamethasone is preferable for patients with suspected but unproved adrenal insufficiency (Addison disease), because the physician can simultaneously treat the patient while performing a diagnostic cosyntropin stimulation test. Methylprednisolone and dexamethasone have negligible mineralocorticoid effects. Large doses of hydrocortisone (ie, even double or triple the stress doses previously mentioned) are preferred if the patient is hypovolemic, hyponatremic, or hyperkalemic, due to the mineralocorticoid effects of hydrocortisone (lacking in prednisone or dexamethasone). No parenteral form of a mineralocorticoid is currently available in the United States. However, if the patient has good gastrointestinal function, fludrocortisone 0. 1 -0. 2 mg may be orally administered.
 • In addition to corticosteroid replacement, aggressive fluid replacement with 5% or 10% intravenous dextrose and saline solutions and treatment of hyperkalemia is mandatory. • A thorough search for a precipitating cause and administration of empiric antibiotics is indicated. Reversal of coagulopathy should be attempted with fresh frozen plasma. • Pressors (eg, dopamine, norepinephrine) may be necessary to combat hypotension.
• In addition to corticosteroid replacement, aggressive fluid replacement with 5% or 10% intravenous dextrose and saline solutions and treatment of hyperkalemia is mandatory. • A thorough search for a precipitating cause and administration of empiric antibiotics is indicated. Reversal of coagulopathy should be attempted with fresh frozen plasma. • Pressors (eg, dopamine, norepinephrine) may be necessary to combat hypotension.
 and human immunodeficiency virus") Do not forget that chronic infections, such as tuberculosis (TB) and human immunodeficiency virus (HIV) infection, can impair adrenal function. The possibility of central adrenal insufficiency (Addison disease) must be investigated, identified, and treated in all patients who have undergone pituitary surgery, irradiation, or prolonged treatment with glucocorticoids.
Do not forget that chronic infections, such as tuberculosis (TB) and human immunodeficiency virus (HIV) infection, can impair adrenal function. The possibility of central adrenal insufficiency (Addison disease) must be investigated, identified, and treated in all patients who have undergone pituitary surgery, irradiation, or prolonged treatment with glucocorticoids.
 due to glucocorticoid therapy can be prevented by giving") Iatrogenic adrenal insufficiency (Addison disease) due to glucocorticoid therapy can be prevented by giving the patient dosages below his or her physiologic requirements. Treatment with alternate -day oral prednisone, or with topical or inhaled glucocorticoids, can reduce the risk of iatrogenic adrenal insufficiency (Addison disease). Glucocorticoid or mineralocorticoid replacement is not contraindicated when needed. This therapy is involved in few drug-drug interactions.
Iatrogenic adrenal insufficiency (Addison disease) due to glucocorticoid therapy can be prevented by giving the patient dosages below his or her physiologic requirements. Treatment with alternate -day oral prednisone, or with topical or inhaled glucocorticoids, can reduce the risk of iatrogenic adrenal insufficiency (Addison disease). Glucocorticoid or mineralocorticoid replacement is not contraindicated when needed. This therapy is involved in few drug-drug interactions.
, long-term glucocorticoid replacement must") Long-Term Monitoring In a child with adrenal insufficiency (Addison disease), long-term glucocorticoid replacement must be balanced between the need to prevent symptoms of adrenal insufficiency (Addison disease) and the need to allow the child to grow at a normal rate, because excess replacement with glucocorticoid diminishes growth velocity. Hydrocortisone is available in 5 -mg, 10 -mg, and 20 mg tablets. This agent is recommended for longterm therapy because of its relatively low potency, which eases the titration of appropriate doses.
Long-Term Monitoring In a child with adrenal insufficiency (Addison disease), long-term glucocorticoid replacement must be balanced between the need to prevent symptoms of adrenal insufficiency (Addison disease) and the need to allow the child to grow at a normal rate, because excess replacement with glucocorticoid diminishes growth velocity. Hydrocortisone is available in 5 -mg, 10 -mg, and 20 mg tablets. This agent is recommended for longterm therapy because of its relatively low potency, which eases the titration of appropriate doses.
 In a large patient, prednisone or dexamethasone may be substituted; however, individual sensitivity to these drugs widely varies. Estimated equivalencies are as follows: • 1 mg of prednisone = typically given as 4 -6 mg of hydrocortisone, but may be up to 15 mg • 1 mg of dexamethasone = 40 -100 mg of hydrocortisone Individualize the maintenance dosage for each patient. The range for hydrocortisone is 7 -20 mg/m 2/d given orally in 2 or 3 divided doses. Patients with primary adrenal insufficiency (Addison disease) who also have mineralocorticoid deficiency require fludrocortisone at 0. 1 -0. 2 mg/d. Young patients must be given adequate access to sodium chloride (typically 2 -4 g/d) to counteract salt wasting.
In a large patient, prednisone or dexamethasone may be substituted; however, individual sensitivity to these drugs widely varies. Estimated equivalencies are as follows: • 1 mg of prednisone = typically given as 4 -6 mg of hydrocortisone, but may be up to 15 mg • 1 mg of dexamethasone = 40 -100 mg of hydrocortisone Individualize the maintenance dosage for each patient. The range for hydrocortisone is 7 -20 mg/m 2/d given orally in 2 or 3 divided doses. Patients with primary adrenal insufficiency (Addison disease) who also have mineralocorticoid deficiency require fludrocortisone at 0. 1 -0. 2 mg/d. Young patients must be given adequate access to sodium chloride (typically 2 -4 g/d) to counteract salt wasting.
.") Medication Summary Glucocorticoid replacement is required in all forms of adrenal insufficiency (Addison disease). Mineralocorticoid replacement is required only in primary adrenal insufficiency (Addison disease), because aldosterone secretion is reduced in primary adrenal insufficiency but not in secondary (central) adrenal insufficiency. In acute adrenal crisis (eg, hypotension, hypoglycemia) use pharmacologic doses of glucocorticoids, which can be in the form of hydrocortisone, methylprednisolone, or dexamethasone.
Medication Summary Glucocorticoid replacement is required in all forms of adrenal insufficiency (Addison disease). Mineralocorticoid replacement is required only in primary adrenal insufficiency (Addison disease), because aldosterone secretion is reduced in primary adrenal insufficiency but not in secondary (central) adrenal insufficiency. In acute adrenal crisis (eg, hypotension, hypoglycemia) use pharmacologic doses of glucocorticoids, which can be in the form of hydrocortisone, methylprednisolone, or dexamethasone.
. The term congenital adrenal hyperplasia (CAH) encompasses a group of") Congenital Adrenal Hyperplasia (CAH). The term congenital adrenal hyperplasia (CAH) encompasses a group of autosomal recessive disorders, each of which involves a deficiency of an enzyme involved in the synthesis of cortisol, aldosterone, or both.
Congenital Adrenal Hyperplasia (CAH). The term congenital adrenal hyperplasia (CAH) encompasses a group of autosomal recessive disorders, each of which involves a deficiency of an enzyme involved in the synthesis of cortisol, aldosterone, or both.
 Pathophysiology The clinical manifestations of each form of congenital adrenal hyperplasia are related to the degree of cortisol deficiency and/or the degree of aldosterone deficiency. In some cases, these manifestations reflect the accumulation of precursor adrenocortical hormones. When present in supraphysiologic concentrations, these precursors lead to excess androgen production with resultant virilization, or because of mineralocorticoid properties, cause sodium retention and hypertension.
Pathophysiology The clinical manifestations of each form of congenital adrenal hyperplasia are related to the degree of cortisol deficiency and/or the degree of aldosterone deficiency. In some cases, these manifestations reflect the accumulation of precursor adrenocortical hormones. When present in supraphysiologic concentrations, these precursors lead to excess androgen production with resultant virilization, or because of mineralocorticoid properties, cause sodium retention and hypertension.
 The phenotype depends on the degree or type of gene deletion or mutation and the resultant deficiency of the steroidogenic enzyme.
The phenotype depends on the degree or type of gene deletion or mutation and the resultant deficiency of the steroidogenic enzyme.
 21 -hydroxylase deficiency: Pathophysiology
21 -hydroxylase deficiency: Pathophysiology
 Frequency The most common form of congenital adrenal hyperplasia is due to mutations or deletions of CYP 21 A, resulting in 21 -hydroxylase deficiency. This deficiency accounts for more than 90% of adrenal hyperplasia cases. Classic adrenal hyperplasia has an overall prevalence of 1 case per 16, 000 population. Congenital adrenal hyperplasia caused by 21 hydroxylase deficiency is found in all populations. 11 -betahydroxylase deficiency is more common in persons of Moroccan or Iranian-Jewish descent.
Frequency The most common form of congenital adrenal hyperplasia is due to mutations or deletions of CYP 21 A, resulting in 21 -hydroxylase deficiency. This deficiency accounts for more than 90% of adrenal hyperplasia cases. Classic adrenal hyperplasia has an overall prevalence of 1 case per 16, 000 population. Congenital adrenal hyperplasia caused by 21 hydroxylase deficiency is found in all populations. 11 -betahydroxylase deficiency is more common in persons of Moroccan or Iranian-Jewish descent.
 The morbidity of the various forms of adrenal hyperplasia is best understood in the context of the steroidogenic pathway used by the adrenal glands and gonads. Severe forms of congenital adrenal hyperplasia are potentially fatal if unrecognized and untreated because of the severe cortisol and aldosterone deficiencies that result in salt wasting, hyponatremia, hyperkalemia, dehydration, and hypotension.
The morbidity of the various forms of adrenal hyperplasia is best understood in the context of the steroidogenic pathway used by the adrenal glands and gonads. Severe forms of congenital adrenal hyperplasia are potentially fatal if unrecognized and untreated because of the severe cortisol and aldosterone deficiencies that result in salt wasting, hyponatremia, hyperkalemia, dehydration, and hypotension.
 Steroidogenic pathway for cortisol, aldosterone, and sex steroid synthesis. A mutation or deletion of any of the genes that code for enzymes involved in cortisol or aldosterone synthesis results in congenital adrenal hyperplasia. The particular phenotype that results depends on the sex of the individual, the location of the block in synthesis, and the severity of the genetic deletion or mutation.
Steroidogenic pathway for cortisol, aldosterone, and sex steroid synthesis. A mutation or deletion of any of the genes that code for enzymes involved in cortisol or aldosterone synthesis results in congenital adrenal hyperplasia. The particular phenotype that results depends on the sex of the individual, the location of the block in synthesis, and the severity of the genetic deletion or mutation.
 Because all forms of congenital adrenal hyperplasia are autosomal recessive disorders, both sexes are affected with equal frequency. Classic congenital adrenal hyperplasia is generally recognized at birth or in early childhood because of ambiguous genitalia, salt wasting, or early virilization. Nonclassic adrenal hyperplasia is generally recognized at or after puberty because of oligomenorrhea or virilizing signs in females.
Because all forms of congenital adrenal hyperplasia are autosomal recessive disorders, both sexes are affected with equal frequency. Classic congenital adrenal hyperplasia is generally recognized at birth or in early childhood because of ambiguous genitalia, salt wasting, or early virilization. Nonclassic adrenal hyperplasia is generally recognized at or after puberty because of oligomenorrhea or virilizing signs in females.
 Clinical presentation in females • Females with severe forms of adrenal hyperplasia due to deficiencies of 21 -hydroxylase, 11 -beta-hydroxylase or 3 -beta-hydroxysteroid dehydrogenase have ambiguous genitalia at birth due to excess adrenal androgen production in utero. This is often called classic virilizing adrenal hyperplasia. • Mild forms of 21 -hydroxylase deficiency in females are identified later in childhood because of precocious pubic hair, clitoromegaly, or both, often accompanied by accelerated growth and skeletal maturation due to excess postnatal exposure to adrenal androgens. This is called simple virilizing adrenal hyperplasia.
Clinical presentation in females • Females with severe forms of adrenal hyperplasia due to deficiencies of 21 -hydroxylase, 11 -beta-hydroxylase or 3 -beta-hydroxysteroid dehydrogenase have ambiguous genitalia at birth due to excess adrenal androgen production in utero. This is often called classic virilizing adrenal hyperplasia. • Mild forms of 21 -hydroxylase deficiency in females are identified later in childhood because of precocious pubic hair, clitoromegaly, or both, often accompanied by accelerated growth and skeletal maturation due to excess postnatal exposure to adrenal androgens. This is called simple virilizing adrenal hyperplasia.
 • Still milder deficiencies of 21 -hydroxylase or 3 beta-hydroxysteroid dehydrogenase activity may present in adolescence or adulthood with oligomenorrhea, hirsutism, and/or infertility. This is termed nonclassic adrenal hyperplasia. • Females with 17 -hydroxylase deficiency appear phenotypically female at birth but do not develop breasts or menstruate in adolescence because of inadequate estradiol production. They may present with hypertension.
• Still milder deficiencies of 21 -hydroxylase or 3 beta-hydroxysteroid dehydrogenase activity may present in adolescence or adulthood with oligomenorrhea, hirsutism, and/or infertility. This is termed nonclassic adrenal hyperplasia. • Females with 17 -hydroxylase deficiency appear phenotypically female at birth but do not develop breasts or menstruate in adolescence because of inadequate estradiol production. They may present with hypertension.
 Clinical presentation in males • 21 -hydroxylase deficiency in males is generally not identified in the neonatal period because the genitalia are normal. If the defect is severe and results in salt wasting, these male neonates present at age 1 -4 weeks with failure to thrive, recurrent vomiting, dehydration, hypotension, hyponatremia, hyperkalemia, and shock (classic salt-wasting adrenal hyperplasia). Patients with less severe deficiencies of 21 -hydroxylase present later in childhood because of the early development of pubic hair, phallic enlargement, or both, accompanied by accelerated linear growth and advancement of skeletal maturation (simple virilizing adrenal hyperplasia).
Clinical presentation in males • 21 -hydroxylase deficiency in males is generally not identified in the neonatal period because the genitalia are normal. If the defect is severe and results in salt wasting, these male neonates present at age 1 -4 weeks with failure to thrive, recurrent vomiting, dehydration, hypotension, hyponatremia, hyperkalemia, and shock (classic salt-wasting adrenal hyperplasia). Patients with less severe deficiencies of 21 -hydroxylase present later in childhood because of the early development of pubic hair, phallic enlargement, or both, accompanied by accelerated linear growth and advancement of skeletal maturation (simple virilizing adrenal hyperplasia).
 • In male infants, the disease may be misdiagnosed as gastroenteritis or pyloric stenosis, with potentially disastrous consequences due to delayed treatment with glucocorticoids. • Males with steroidogenic acute regulatory (St. AR) deficiency, classic 3 -beta-hydroxysteroid dehydrogenase deficiency, or 17 -hydroxylase deficiency generally have ambiguous genitalia or female genitalia because of inadequate testosterone production in the first trimester of fetal life.
• In male infants, the disease may be misdiagnosed as gastroenteritis or pyloric stenosis, with potentially disastrous consequences due to delayed treatment with glucocorticoids. • Males with steroidogenic acute regulatory (St. AR) deficiency, classic 3 -beta-hydroxysteroid dehydrogenase deficiency, or 17 -hydroxylase deficiency generally have ambiguous genitalia or female genitalia because of inadequate testosterone production in the first trimester of fetal life.
 Other findings • Hyponatremia, hyperkalemia, and/or hypoglycemia suggests the possibility of adrenal insufficiency. • Hypoglycemia and hypotension may, in part, be due to associated epinephrine synthesis in the adrenal medulla due to cortisol deficiency. Cortisol, perfusing the adrenal medulla from the cortex, normally stimulates phenylethanolamine N -methyltransferase, the last enzyme in epinephrine synthesis. • Children with simple virilizing 21 -hydroxylase deficiency or 11 hydroxylase deficiency have early pubic hair, phallic enlargement, and accelerated linear growth and advanced skeletal maturation. • Two forms of adrenal hyperplasia (ie, 11 -hydroxylase [CYP 11 B 1] and 17 -hydroxylase [CYP 17] deficiency) result in hypertension due to the accumulation of supraphysiologic concentrations of deoxycorticosterone. • Patients with aldosterone deficiency of any etiology may present with dehydration, hyponatremia, and hyperkalemia, especially with the stress of illness.
Other findings • Hyponatremia, hyperkalemia, and/or hypoglycemia suggests the possibility of adrenal insufficiency. • Hypoglycemia and hypotension may, in part, be due to associated epinephrine synthesis in the adrenal medulla due to cortisol deficiency. Cortisol, perfusing the adrenal medulla from the cortex, normally stimulates phenylethanolamine N -methyltransferase, the last enzyme in epinephrine synthesis. • Children with simple virilizing 21 -hydroxylase deficiency or 11 hydroxylase deficiency have early pubic hair, phallic enlargement, and accelerated linear growth and advanced skeletal maturation. • Two forms of adrenal hyperplasia (ie, 11 -hydroxylase [CYP 11 B 1] and 17 -hydroxylase [CYP 17] deficiency) result in hypertension due to the accumulation of supraphysiologic concentrations of deoxycorticosterone. • Patients with aldosterone deficiency of any etiology may present with dehydration, hyponatremia, and hyperkalemia, especially with the stress of illness.
 • Male or female patients with 11 -hydroxylase deficiency may present in the second or third week of life with a salt-losing crisis. However, these patients develop hypertension, hypokalemic alkalosis, or both later in life. This paradox is explained by resistance to mineralocorticoids in infancy and the inability of the elevated deoxycorticosterone levels to replace the deficient serum concentrations of aldosterone in infancy. Upon maturation, mineralocorticoid responsiveness increases, and the elevated concentrations of deoxycorticosterone are sufficient to cause sodium retention, potassium excretion, and hypertension.
• Male or female patients with 11 -hydroxylase deficiency may present in the second or third week of life with a salt-losing crisis. However, these patients develop hypertension, hypokalemic alkalosis, or both later in life. This paradox is explained by resistance to mineralocorticoids in infancy and the inability of the elevated deoxycorticosterone levels to replace the deficient serum concentrations of aldosterone in infancy. Upon maturation, mineralocorticoid responsiveness increases, and the elevated concentrations of deoxycorticosterone are sufficient to cause sodium retention, potassium excretion, and hypertension.
 Differential Diagnoses 3 -Beta-Hydroxysteroid Dehydrogenase Deficiency; 5 -Alpha-Reductase Deficiency; Adrenal Hypoplasia; Adrenal Insufficiency; Ambiguous Genitalia and Intersexuality; Androgen Insensitivity Syndrome; Congenital Adrenal Hyperplasia; Cystic Fibrosis; Denys-Drash Syndrome; Failure to Thrive. Familial Glucocorticoid Deficiency; Fluid, Electrolyte, and Nutrition Management of the Newborn; Hyperkalemia; Hyponatremia; Pyloric Stenosis, Hypertrophic; Sexuality: Gender Identity; Sexuality: Sexual Orientation; Small-Bowel Obstruction; WAGR Syndrome (Wilms tumor, Aniridia, Genitourinary defect, mental Retardation complex).
Differential Diagnoses 3 -Beta-Hydroxysteroid Dehydrogenase Deficiency; 5 -Alpha-Reductase Deficiency; Adrenal Hypoplasia; Adrenal Insufficiency; Ambiguous Genitalia and Intersexuality; Androgen Insensitivity Syndrome; Congenital Adrenal Hyperplasia; Cystic Fibrosis; Denys-Drash Syndrome; Failure to Thrive. Familial Glucocorticoid Deficiency; Fluid, Electrolyte, and Nutrition Management of the Newborn; Hyperkalemia; Hyponatremia; Pyloric Stenosis, Hypertrophic; Sexuality: Gender Identity; Sexuality: Sexual Orientation; Small-Bowel Obstruction; WAGR Syndrome (Wilms tumor, Aniridia, Genitourinary defect, mental Retardation complex).
 Laboratory Studies The diagnosis of congenital adrenal hyperplasia depends on the demonstration of inadequate production of cortisol, aldosterone, or both in the presence of accumulation of excess concentrations of precursor hormones.
Laboratory Studies The diagnosis of congenital adrenal hyperplasia depends on the demonstration of inadequate production of cortisol, aldosterone, or both in the presence of accumulation of excess concentrations of precursor hormones.
 The distinguishing characteristic of 21 hydroxylase deficiency is a high serum concentration of 17 -hydroxyprogesterone (usually >1000 ng/d. L) and urinary pregnanetriol (metabolite of 17 -hydroxyprogesterone) in the presence of clinical features suggestive of the disease (eg, salt wasting, clitoromegaly or ambiguous genitalia, precocious pubic hair, excessive growth, premature phallic enlargement in the absence of testicular enlargement, hirsutism, oligomenorrhea, female infertility).
The distinguishing characteristic of 21 hydroxylase deficiency is a high serum concentration of 17 -hydroxyprogesterone (usually >1000 ng/d. L) and urinary pregnanetriol (metabolite of 17 -hydroxyprogesterone) in the presence of clinical features suggestive of the disease (eg, salt wasting, clitoromegaly or ambiguous genitalia, precocious pubic hair, excessive growth, premature phallic enlargement in the absence of testicular enlargement, hirsutism, oligomenorrhea, female infertility).
 The 11 -beta-hydroxylase deficiency is indicated by excess concentrations of 11 -deoxycortisol and deoxycorticosterone or by an elevation in the ratio of 24 -hour urinary tetrahydrocompound S (metabolite of 11 deoxycortisol) to tetrahydrocompound F (metabolite of cortisol). Both forms of adrenal hyperplasia are accompanied by elevated levels of 24 -hour urinary 17 -ketosteroids, the urinary metabolites of adrenal androgens.
The 11 -beta-hydroxylase deficiency is indicated by excess concentrations of 11 -deoxycortisol and deoxycorticosterone or by an elevation in the ratio of 24 -hour urinary tetrahydrocompound S (metabolite of 11 deoxycortisol) to tetrahydrocompound F (metabolite of cortisol). Both forms of adrenal hyperplasia are accompanied by elevated levels of 24 -hour urinary 17 -ketosteroids, the urinary metabolites of adrenal androgens.
 • 3 -beta-hydroxysteroid dehydrogenase deficiency is indicated by an abnormal ratio of 17 hydroxypregnenolone to 17 hydroxyprogesterone and dehydroepiandrosterone to androstenedione. • Salt-wasting forms of adrenal hyperplasia are accompanied by low serum aldosterone concentrations, hyponatremia, hyperkalemia, and elevated plasma renin activity (PRA), indicating hypovolemia. In contrast, hypertensive forms of adrenal hyperplasia (i. e. , 11 -betahydroxylase deficiency and 17 -alpha-hydroxylase deficiency) are associated with suppressed PRA and, often, hypokalemia.
• 3 -beta-hydroxysteroid dehydrogenase deficiency is indicated by an abnormal ratio of 17 hydroxypregnenolone to 17 hydroxyprogesterone and dehydroepiandrosterone to androstenedione. • Salt-wasting forms of adrenal hyperplasia are accompanied by low serum aldosterone concentrations, hyponatremia, hyperkalemia, and elevated plasma renin activity (PRA), indicating hypovolemia. In contrast, hypertensive forms of adrenal hyperplasia (i. e. , 11 -betahydroxylase deficiency and 17 -alpha-hydroxylase deficiency) are associated with suppressed PRA and, often, hypokalemia.
 Imaging Studies • Imaging studies of the adrenal gland are generally not useful in the evaluation of patients with suspected adrenal hyperplasia. However, CT scanning of the adrenal gland can be useful in excluding bilateral adrenal hemorrhage in patients with signs of acute adrenal failure without ambiguous genitalia or other clues of adrenal hyperplasia. • Pelvic ultrasonography may be performed in an infant with ambiguous genitalia to demonstrate a uterus or associated renal anomalies, which are sometimes found in other conditions that may result in ambiguous genitalia (eg, mixed gonadal dysgenesis, Denys-Drash syndrome). • Urogenitography is often helpful in defining the anatomy of the internal genitalia. • A bone-age study is useful in evaluating a child who develops precocious pubic hair, clitoromegaly, or accelerated linear growth. Patients who have these symptoms because of adrenal hyperplasia have advanced skeletal maturation.
Imaging Studies • Imaging studies of the adrenal gland are generally not useful in the evaluation of patients with suspected adrenal hyperplasia. However, CT scanning of the adrenal gland can be useful in excluding bilateral adrenal hemorrhage in patients with signs of acute adrenal failure without ambiguous genitalia or other clues of adrenal hyperplasia. • Pelvic ultrasonography may be performed in an infant with ambiguous genitalia to demonstrate a uterus or associated renal anomalies, which are sometimes found in other conditions that may result in ambiguous genitalia (eg, mixed gonadal dysgenesis, Denys-Drash syndrome). • Urogenitography is often helpful in defining the anatomy of the internal genitalia. • A bone-age study is useful in evaluating a child who develops precocious pubic hair, clitoromegaly, or accelerated linear growth. Patients who have these symptoms because of adrenal hyperplasia have advanced skeletal maturation.
 Medical Care • Patients with dehydration, hyponatremia, or hyperkalemia and a possible salt-wasting form of adrenal hyperplasia should receive an intravenous (IV) bolus of isotonic sodium chloride solution (20 m. L/kg or 450 m. L/m 2) over the first hour, as needed, to restore their intravascular volume and blood pressure. This dosage may be repeated if the blood pressure remains low. • Dextrose must be administered if the patient is hypoglycemic and must be included in the rehydration fluid after the bolus dose to prevent hypoglycemia.
Medical Care • Patients with dehydration, hyponatremia, or hyperkalemia and a possible salt-wasting form of adrenal hyperplasia should receive an intravenous (IV) bolus of isotonic sodium chloride solution (20 m. L/kg or 450 m. L/m 2) over the first hour, as needed, to restore their intravascular volume and blood pressure. This dosage may be repeated if the blood pressure remains low. • Dextrose must be administered if the patient is hypoglycemic and must be included in the rehydration fluid after the bolus dose to prevent hypoglycemia.
 • After the patient's condition is stabilized, treat all patients who have adrenal hyperplasia with longterm glucocorticoid or aldosterone replacement (or both), depending on which enzyme is involved and on whether cortisol and/or aldosterone synthesis is affected. • Some patients develop precocious puberty, which further compromises adult height. Suppression of puberty with long-acting gonadotropin-releasing hormone (Gn. RH) agonists while simultaneously stimulating growth with growth hormone may partially improve the patient's height
• After the patient's condition is stabilized, treat all patients who have adrenal hyperplasia with longterm glucocorticoid or aldosterone replacement (or both), depending on which enzyme is involved and on whether cortisol and/or aldosterone synthesis is affected. • Some patients develop precocious puberty, which further compromises adult height. Suppression of puberty with long-acting gonadotropin-releasing hormone (Gn. RH) agonists while simultaneously stimulating growth with growth hormone may partially improve the patient's height
 Long-term medical therapy The goal of therapy for adrenal hyperplasia is the replacement of glucocorticoid and mineralocorticoid to prevent signs of adrenal insufficiency and to prevent the accumulation of precursor hormones that cause virilization. The dosage must be tailored to each patient, but the general average dosage is 10 -25 mg/m 2/d of hydrocortisone PO divided in 2 -3 doses. Hydrocortisone is recommended in the pediatric population because of its lower potency, which permits easier titration of appropriate doses.
Long-term medical therapy The goal of therapy for adrenal hyperplasia is the replacement of glucocorticoid and mineralocorticoid to prevent signs of adrenal insufficiency and to prevent the accumulation of precursor hormones that cause virilization. The dosage must be tailored to each patient, but the general average dosage is 10 -25 mg/m 2/d of hydrocortisone PO divided in 2 -3 doses. Hydrocortisone is recommended in the pediatric population because of its lower potency, which permits easier titration of appropriate doses.
 Prednisone, prednisolone, or even dexamethasone suspensions may be used. Prednisone is available in a suspension of 1 mg/m. L, and prednisolone is available in a solution of 5 or 15 mg/5 m. L. The estimated equivalencies are as follows: • One mg of prednisone is equal to 4 mg of hydrocortisone. • One mg of prednisolone is equal to 5 mg of hydrocortisone. • One mg of dexamethasone is equal to 50 mg of hydrocortisone. Administer fludrocortisone (0. 05 -0. 2 mg/d PO) to patients with mineralocorticoid deficiency.
Prednisone, prednisolone, or even dexamethasone suspensions may be used. Prednisone is available in a suspension of 1 mg/m. L, and prednisolone is available in a solution of 5 or 15 mg/5 m. L. The estimated equivalencies are as follows: • One mg of prednisone is equal to 4 mg of hydrocortisone. • One mg of prednisolone is equal to 5 mg of hydrocortisone. • One mg of dexamethasone is equal to 50 mg of hydrocortisone. Administer fludrocortisone (0. 05 -0. 2 mg/d PO) to patients with mineralocorticoid deficiency.
 The Endocrine Society's 2010 clinical practice guidelines note the following: • Prenatal treatment for CAH should be regarded as experimental. • Glucocorticoid therapy should be carefully titrated to avoid Cushing syndrome. • Mineralocorticoid replacement is encouraged. In infants, mineralocorticoid replacement and sodium supplementation are encouraged. • Use of agents to delay puberty and promote growth are experimental. • Psychiatric support should be encouraged for patients with adjustment problems. • Medication should be used judiciously during pregnancy and in symptomatic patients with nonclassical CAH. • Adrenalectomy should be avoided. • While surgical reconstruction may not be necessary during the newborn period in mildly virilized girls, it may be appropriate in severely virilized girls. It should be a single stage genital repair, performed by experienced surgeons.
The Endocrine Society's 2010 clinical practice guidelines note the following: • Prenatal treatment for CAH should be regarded as experimental. • Glucocorticoid therapy should be carefully titrated to avoid Cushing syndrome. • Mineralocorticoid replacement is encouraged. In infants, mineralocorticoid replacement and sodium supplementation are encouraged. • Use of agents to delay puberty and promote growth are experimental. • Psychiatric support should be encouraged for patients with adjustment problems. • Medication should be used judiciously during pregnancy and in symptomatic patients with nonclassical CAH. • Adrenalectomy should be avoided. • While surgical reconstruction may not be necessary during the newborn period in mildly virilized girls, it may be appropriate in severely virilized girls. It should be a single stage genital repair, performed by experienced surgeons.
 Diet and Activity • Patients with congenital adrenal hyperplasia should be on an unrestricted diet. • Patients should have ample access to salt because salt wasting is common in some forms of the disease. • Infants who have salt wasting generally benefit from supplementation with Na. Cl (2 -4 g/d) added to their formula. • Caloric intake may need to be monitored and restricted if excess weight gain occurs because glucocorticoids stimulate appetite. • Activity restriction is not necessary if appropriate glucocorticoid and mineralocorticoid therapy is provided.
Diet and Activity • Patients with congenital adrenal hyperplasia should be on an unrestricted diet. • Patients should have ample access to salt because salt wasting is common in some forms of the disease. • Infants who have salt wasting generally benefit from supplementation with Na. Cl (2 -4 g/d) added to their formula. • Caloric intake may need to be monitored and restricted if excess weight gain occurs because glucocorticoids stimulate appetite. • Activity restriction is not necessary if appropriate glucocorticoid and mineralocorticoid therapy is provided.
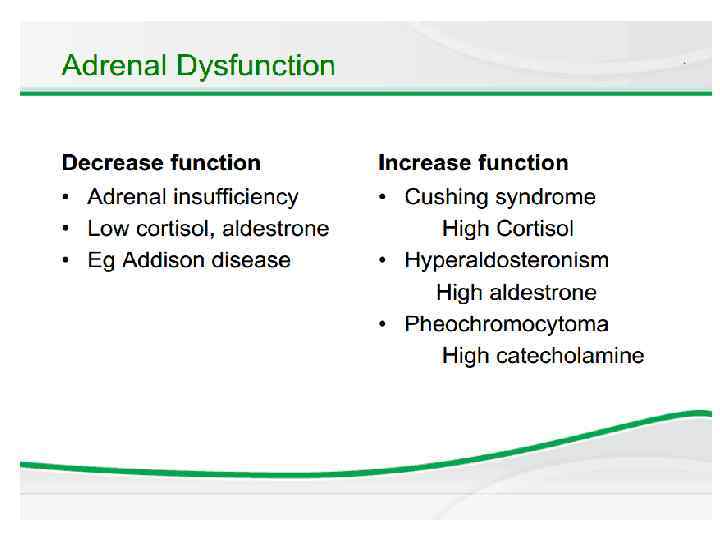
 Adrenal Insufficiency Summary • • May be primary or secondary May be congenital or acquired Treatment is relatively simple Diagnosis is often controversial – Baseline cortisol/ACTH before steroids – ACTH stim test if possible – Additional testing if CAH is suspected • Don’t forget to check the blood sugar!
Adrenal Insufficiency Summary • • May be primary or secondary May be congenital or acquired Treatment is relatively simple Diagnosis is often controversial – Baseline cortisol/ACTH before steroids – ACTH stim test if possible – Additional testing if CAH is suspected • Don’t forget to check the blood sugar!
 Cushing syndrome • Glucocorticoid excess in children is usually a sideeffect of long-term glucocorticoid treatment (intravenous, oral or, more rarely, inhaled, nasal or topical) for conditions such as the nephrotic syndrome, asthma or, in the past, for severe bronchopulmonary dysplasia. • Other causes of glucocorticoid excess are rare. It may be ACTH-driven, from a pituitary adenoma, usually in older children, or from ectopic ACTHproducing tumours, but these almost never occur in children.
Cushing syndrome • Glucocorticoid excess in children is usually a sideeffect of long-term glucocorticoid treatment (intravenous, oral or, more rarely, inhaled, nasal or topical) for conditions such as the nephrotic syndrome, asthma or, in the past, for severe bronchopulmonary dysplasia. • Other causes of glucocorticoid excess are rare. It may be ACTH-driven, from a pituitary adenoma, usually in older children, or from ectopic ACTHproducing tumours, but these almost never occur in children.
 Clinical features of Cushing syndrome • • • Growth failure/short stature Face and trunk obesity Red cheeks Hirsutism Striae Hypertension Bruising Carbohydrate intolerance Muscle wasting and weakness Osteopenia Psychological problems.
Clinical features of Cushing syndrome • • • Growth failure/short stature Face and trunk obesity Red cheeks Hirsutism Striae Hypertension Bruising Carbohydrate intolerance Muscle wasting and weakness Osteopenia Psychological problems.
 Adrenal tumours are identified on CT or MRI scan of the abdomen and a pituitary adenoma on MRI brain scan. Adrenal tumours are usually unilateral and are treated by adrenalectomy and radiotherapy if indicated. Pituitary adenomas are best treated by trans-sphenoidal resection, but radiotherapy can be used.
Adrenal tumours are identified on CT or MRI scan of the abdomen and a pituitary adenoma on MRI brain scan. Adrenal tumours are usually unilateral and are treated by adrenalectomy and radiotherapy if indicated. Pituitary adenomas are best treated by trans-sphenoidal resection, but radiotherapy can be used.