4d36ba4410f4aa353cdcf7cebe1abcfa.ppt
- Количество слайдов: 43




Conditions characterised by abnormal proliferation of leucopoietic tissue and appearance of immature forms of white cells in the peripheral blood.

Acute Leukemias Results from proliferation of young forms of leucocytes at a stage when they do not enter the circulation as readily as they do when more mature.

Classification: * acute lymphoblastic L. > more common * acute myeloblastic L. * acute monocytic L.

ACUTE LEUKEMIAS Def: Heterogenous group of neoplastic discases characterized. by proliferation of atypical elements which originates from the stem cells of the hematopoietic system. the uncontrolled and progressive proliferation of these cells lead to: Replacement of normal marrow Invasion of peripheral blood Infiltration of various organs and tissues
 Adults >14 years % % Lymphoblastic")
Relative frequency : Type Children (≤ 14 years) Adults >14 years % % Lymphoblastic 65 80 10 25 Myeloblastic 6 25 20 Myelomonocytic 2 6 20 Monocytic 2 6 1 10 Promyelocytic 1 2 10 Erythroid 0 1 3 Megakaryocytic 0 1 1

Etiology: Unknown, some factors seem to be implicated: A Environmental factors: • Ionizing radiation; mainly AML but ALL less important • Chemical substances; prolonged exposure to certain substances (benzene, phenylbutazone, chloramphenicol, anticancer drugs, alkylating agents, natulan) > incidence of leukemia mainly AML Onset often preceeded by a state of bone marrow hypoplasia, peripheral pancytopenia (preleukemic syndrome).

B Genetic: May act by facilitating envirnomental factors. C Viruses: Based on experiments with laboratory animal, has never proved humans. Typical products of viruses have been identified in some adult patients. With T. ALL: HTLV (human T cell lymphotrophic virus) in T cell ALL and hairy cell leuk.

D Immunological Factors: Patient with acquired or congenital immune deficiency syndrome or subjected to prolonged immunosuppresive treatment has ↑ incidence of leukemia.

Differential Diagnosis : D. D. between various forms of acute leukemia is mainly on cytomorphologic, cytochemical criteria rather than on clinical data, which are often superimposable. age: may accur at any age but in general: A. L. L > the peak incidence in 1 st 6 years of life. Uncommon after age of 20 A. M. L. >more commonly at slightly older age groups centering around the teen age or adolescent period A. Monocytic leuk. >no specific age group.

Clinical Picture 1 Fever: moderate or high grade, usually irregular and shoots up when 2 ry infection occur 2 rapidly developing marked asthenia. 3 rapidly developing severe anaemia > marked pallor. 4 severe bone ache allover the body 5 Severe sore throat 6 bleeding tendency from > > gums. > skin > orifices >int. organs. 7 tender bones 8 joint pain (bleeding & infiltration) 9 L. N. enlargement in > > A. L. L. >A. M. L. >A. Monocytic L. 10 spleen & Liver enlargement. 11 Chloromas : - more in A. M. L. (subperiosteal tumor like masses) 12 C. N. S >Focal Hge or infection of nerves or meningies.

Blood Picture : A. L. L : * R. B. Cs > severe form of normocytic ncrmochromic anemia occurs early. * W. B. cs > ( ) 10, 000 100, 000 Or more but 40% are leukopenic Lymphocytes are likely to be the predominant cells. The diagnosis is established by detecting lymphoblastes in the peripheral smear. (N. B. Lymphoblastes : large cells with clear cytoplasm, prominant nucleus, definite nucleole. Diff. From mycloblastes : absent specific granules of cytoplasm, ve peroxidase stain PAS +ve Sudan Black ve or weekly +ve * platelets : usually < 100. 000 may be completetly absent.

2 Myeloblastic Leuk W. B. Cs. count as A. L. L. Leukopenia is as likely to occur as A. L. L The cells are peroxidase +ve, PAS –ve, Sudan black +ve , acid phosphatase +ve In 10 20% : Auer bodies are present rod like structures. in cytoplasm of > myeloblast > monoblast 3 Monocytic Leukemia: monoblast in the peripheral blood. Stain PAS ve, Sudan B +ve, Peroxidase ve acid phosphatase +ve
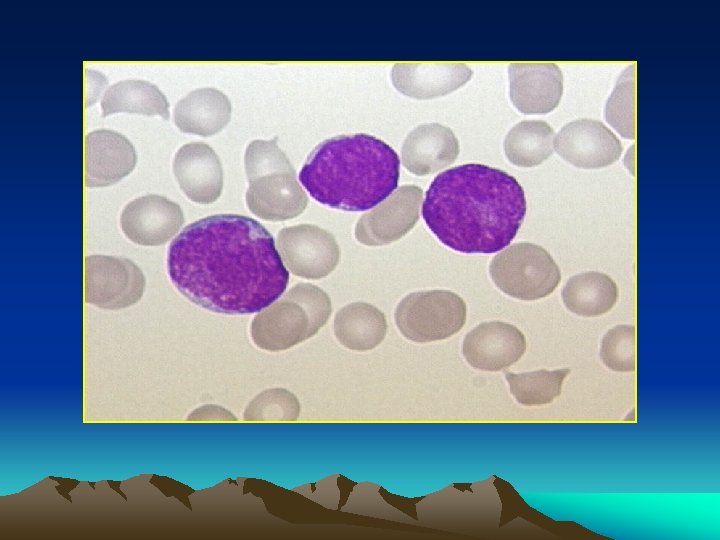
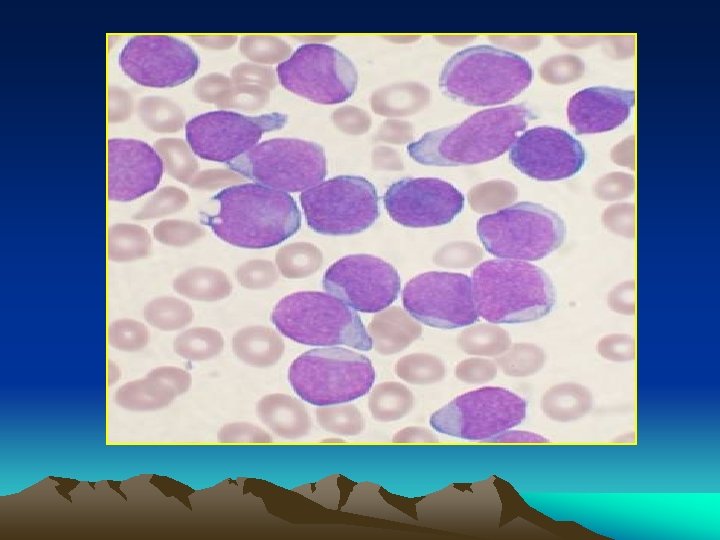
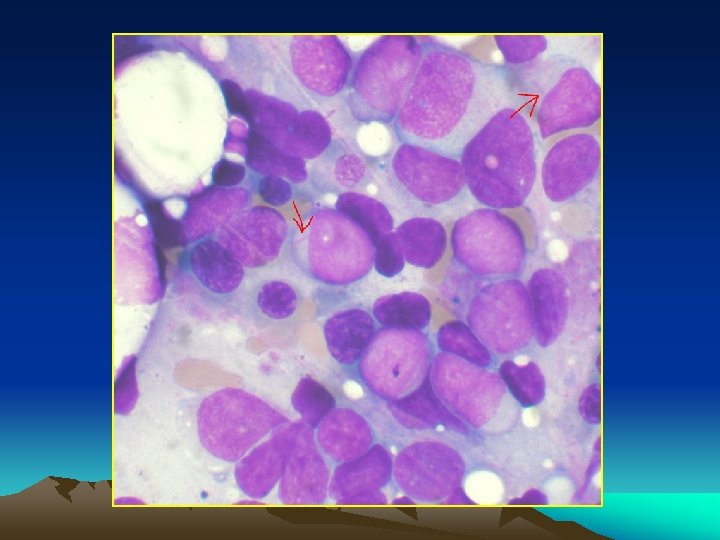

Bone marrow aspiration: Massive proliferation of blast cells even when leukopenia exist. * A. L. L > lymphoblast * A. M. L >myeloblast *A. monocytic > monoblasts and monocytes. *Radiology : Sketetat involvement in almost all children and 50% of dults. * diffuse osteoprosis * periosteal elevation * osteolytic lesions * radiolucent metaphyseal bands.
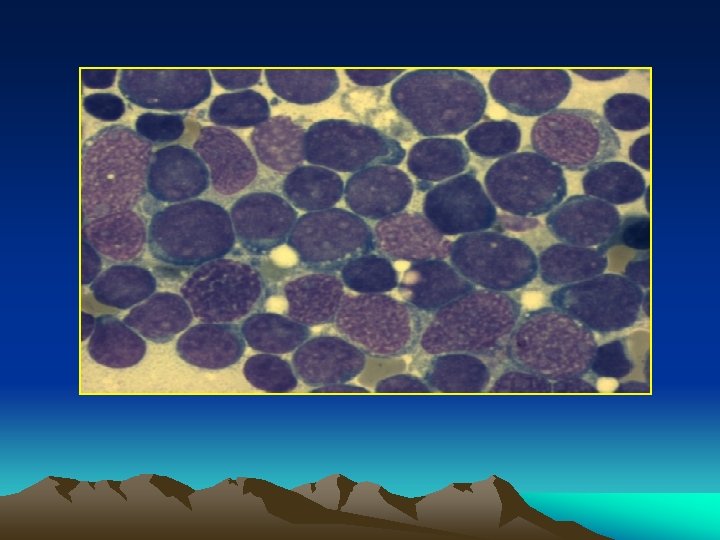

Diff diagnosis : The combination of * anemia * thrombocytopenia * bone marrow prolif. è primitive white cells is found only in leukemia.
From other causes of sore throat + Fever as: * vincent angina. *diphtheria. *infectious,")
(1)From other causes of sore throat + Fever as: * vincent angina. *diphtheria. *infectious, mononucleosis. *aplastic anemia. *agranulocytosis. (2) from other causes of parpura : * I. T. P *Aplastic anemia.
 Lymphadenopathy + splenomegally: * infective mononucleosis *H. D. . N. H. L. (by")
(3) Lymphadenopathy + splenomegally: * infective mononucleosis *H. D. . N. H. L. (by blood picture). . (4) Lymphocytosis : in : . * whooping caugh *infective lyrmphocytosis. (white cells mature, R. B. Cs and platelets are normal). (5) Rheumtic Fever
![Name Description • Includes: AML with translocations between chromosome 8 and 21 [t(8; 21)]](https://present5.com/presentation/4d36ba4410f4aa353cdcf7cebe1abcfa/image-22.jpg "Name Description • Includes: AML with translocations between chromosome 8 and 21 [t(8; 21)]")
Name Description • Includes: AML with translocations between chromosome 8 and 21 [t(8; 21)] (ICD-O 9896/3); RUNX 1/RUNX 1 T 1 • AML with inversions in chromosome 16 [inv(16)] (ICD-O 9871/3); AML with characteristic genetic CBFB/MYH 11 • AML with translocations between abnormalities chromosome 15 and 17 [t(15; 17)] (ICD-O 9866/3); RARA; PML Patients with AML in this category generally have a high rate of remission and a better prognosis compared to other types of AML. This category includes patients who have had a prior myelodysplastic syndrome (MDS) or myeloproliferative AML with multilineage dysplasia disease (MPD) that transforms into AML. This category of AML occurs most often in elderly patients and often has a worse prognosis. This category includes patients who have had prior chemotherapy and/or radiation and subsequently develop AML and MDS, therapy-related AML or MDS. These leukemias may be characterized by specific chromosomal abnormalities, and often carry a worse prognosis. Includes subtypes of AML that do not AML not otherwise categorized fall into the above categories. ICD-O Multiple M 9895/3 M 9920/3 M 9861/3

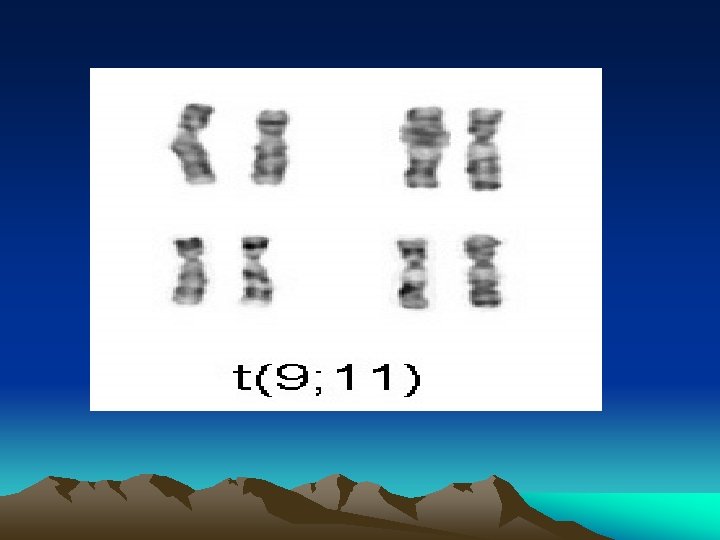
aaa Name Cytogenetics M 0 minimally differentiated acute myeloblastic leukemia M 1 acute myeloblastic leukemia, without maturation M 2 acute myeloblastic leukemia, with granulocytic maturation t(8; 21)(q 22; q 22), t(6; 9) M 3 promyelocytic, or acute promyelocytic leukemia (APL) t(15; 17) M 4 acute myelomonocytic leukemia inv(16)(p 13 q 22), del(16 q) M 4 eo myelomonocytic together with bone marrow eosinophilia inv(16), t(16; 16) M 5 acute monoblastic leukemia (M 5 a) or acute monocytic leukemia (M 5 b) M 6 acute erythroid leukemias, including erythroleukemia (M 6 a) and very rare pure erythroid leukemia (M 6 b) M 7 M 8 acute megakaryoblastic leukemia acute basophilic leukemia del (11 q), t(9; 11), t(11; 19) t(1; 22)

The FAB classification. Subtyping of the various forms of ALL used to be done according to the French-American. British (FAB) classification, [18] which was used for all acute leukemias (including acute myelogenous leukemia, AML). ALL-L 1: small uniform cells ALL-L 2: large varied cells ALL-L 3: large varied cells with vacuoles (bubble-like features) Each subtype is then further classified by determining the surface markers of the abnormal lymphocytes, called immunophenotyping. There are 2 main immunologic types: pre-B cell and pre-T cell. The mature B-cell ALL (L 3) is now classified as Burkitt's lymphoma/leukemia. Subtyping helps determine the prognosis and most appropriate treatment in treating ALL

WHO proposed classification of acute lymphoblastic leukemia The recent WHO International panel on ALL recommends that the FAB classification be abandoned, since the morphological classification has no clinical or prognostic relevance. It instead advocates the use of the immunophenotypic classification mentioned below. 1 - Acute lymphoblastic leukemia/lymphoma Synonyms: Former Fab L 1/L 2 i. Precursor B acute lymphoblastic leukemia/lymphoma. Cytogenetic subtypes: [19] t(12; 21)(p 12, q 22) TEL/AML-1 t(1; 19)(q 23; p 13) PBX/E 2 A t(9; 22)(q 34; q 11) ABL/BCR T(V, 11)(V; q 23) V/MLL ii. Precursor T acute lymphoblastic leukemia/lymphoma 2 - Burkitt's leukemia/lymphoma Synonyms: Former FAB L 3 3 - Biphenotypic acute leukemia

Treatment: Combination chemotherapy in order to : * obtain synergestic action * minimize side effects. * attacks leukemic cells in different phases of mitosis. * delay the onset of resistance of the malignant cells.

Acute L. Leuk. effective drugs are 1 - vincristine-----> arrest cell mitosis 2 - predinsone ----> Lyrmpholysis 3 -6. M. P. ----> inhibit DNA synthesis. 4 -Methotrexate ----> inhibit RNA and protein synthesis 5 -Doxorubich (adriamycin)----> inhibit DNA synthesis 6 -L- asparaginase

Phase Description Agents The aim of remission induction is to rapidly kill most tumor cells and Combination of get the patient into Prednisolone or remission. This is dexamethasone defined as the (in children), presence of less vincristine, Remission than 5% leukemic asparaginase, and induction blasts in the bone daunorubicin marrow, normal (used in Adult blood cells and ALL) is used to absence of tumor induce remission cells from blood, and absence of other signs and symptoms of the disease.

Intensification uses high doses of intravenous multidrug chemotherapy to further reduce tumor burden. Since ALL cells sometimes penetrate the Central Nervous System (CNS), most protocols include delivery of chemotherapy into the CNS fluid (termed intrathecal chemotherapy). Some centers deliver the drug through Ommaya reservoir (a device surgically placed under the scalp and used to deliver drugs to the CNS fluid and to extract CNS fluid for various tests). Other centers would perform multiple lumbar punctures as needed for testing and treatment delivery. Typical intensification protocols use vincristine, cyclophosphamide, cytarabine, daunorubicin, etoposide, thioguanine or mercaptopurine given as blocks in different combinations. For CNS protection, intrathecal methotrexate or cytarabine is usually used combined with or without cranio-spinal irradiation (the use of radiation therapy to the head and spine). Central nervous system relapse is treated with intrathecal administration of hydrocortisone, methotrexate, and cytarabine.

Maintenance therapy The aim of maintenance therapy is to kill any residual cell that was not killed by remission induction, and intensification regimens. Although such cells are few, they will cause relapse if not eradicated. For this purpose, daily oral mercaptopurine, once weekly oral methotrexate, once monthly 5 -day course of intravenous vincristine and oral corticosteroids are usually used. The length of maintenance therapy is 3 years for boys, 2 years for girls and adults.

These drugs must be used sequentially and in combinations: a Induction therapy: * To obtain apparent clinical & hematological remission * 2 or 3 drugs used Vincristine 1. 4 mg/m 2 I. V. once weekly for 4 weeks. . . prednisone lmg/kg body weight P. O. daily L. asparaginase or Adriamycin

b Consolidation theraby: *to decrease total no. of residual malignant cells to 10 G cells or less. * e. g. Methotrexate : 15 mg/m 2 I. M. daily For 3 5 days. . followed by cystarabine 100 mg/ m 2 I. V. twice daily for 3 5 days.

c C. N. S. prophylaxis by * cranial irradiation 1800 2400 R * Intra thecal (l. T. ) Methotrexate in mg/ m 2 for 5 injection. (twice weekly).

d Prolonged maintenance : ~ * 2 3 years * Methotrexate 15 mg/ m 2 once or twice weekly I. M. * 6 M. P 1 2. 5 mg/kg daily P. O. *Cyclophosphamide: 200 mg/ m 2 p. o. weekly.

Acute Non Lymphoblastic L. The initial objectives is the induction of a CR. (reduction of marrow blasts to less than 5%, increase in neutrophils in the peripheral blood to l. 5 x 106 /L. or more and of platelets to 100 x 106/L. or more. ). Then consolidation therapy follows, for which identical drugs are used (the aim is further reduction of leukemic cells). Both in induction and consolidation, high dosage of drugs are given to produce temporary marrow aplasia.

-After the marrow have recovered from consolidation therapy, maintenance therapy is given at monthly intervals usually for 2 -3 years, aiming for a further stepwise reduction of remaining leukemic cells without making the marrow splastic The cornerstone of remission induction and consolidation chemotherapy is a combination of cytosine-arabinoside and daunorubicin resulting in 50% CR rates. Drugs used for maintenance: cytosine-arabinoside, 6 thioguanine, cyclophosphamide and daunarubicin. However the value of maintenance treatment is marjinal.
Severe bone ache or massive C. N. S infiltration. > Local irradiation")
complications : (1)Severe bone ache or massive C. N. S infiltration. > Local irradiation (2)C. N. S. involvement: > intrathecal Mtx. 12 mg I. T. /3 days + cranial irradiation (3)Fever. (4)Hge. (6)Hyperuricemia
 A. L. L >C. R. > 90% of children. ----> 60% of")
Prognosis (1) A. L. L >C. R. > 90% of children. ----> 60% of adults. ----> Cure in 70% of children. and 35% of adults. Allogenic bone marrow transplantation is the only form of therapy for patients with drug -resistant disease (2) A. N. L. L. >C. R is up to 50% 5% only about be cured However, bone marrow transplantation if preformed during First remission can improve survival and it has been suggested to be preformed in all patients younger than 50 years and who have available HLA identical donor
, t(15; 17), inv(16)")
Risk Category Abnormality 5 -year survival Relapse rate Favorable t(8; 21), t(15; 17), inv(16) 70% 33% Intermediate Normal, +8, +21, +22, del(7 q), del(9 q), Abnormal 11 q 23, all other structural or numerical changes 48% 50% Adverse -5, -7, del(5 q), Abnormal 3 q, Complex cytogenetics 15% 78%
![[edit] Prognosis The survival rate has improved from zero four decades ago, to 20](https://present5.com/presentation/4d36ba4410f4aa353cdcf7cebe1abcfa/image-41.jpg "[edit] Prognosis The survival rate has improved from zero four decades ago, to 20")
[edit] Prognosis The survival rate has improved from zero four decades ago, to 20 -75 percent currently, largely due to clinical trials and improvements in bone marrow transplantation (BMT) and stem cell transplantation (SCT) technology. Five-year survival rates evaluate older, not current, treatments. New drugs, and matching treatment to the genetic characteristics of the blast cells, may improve those rates. The prognosis for ALL differs between individuals depending on a variety of factors: Sex: females tend to fare better than males. Ethnicity: Caucasians are more likely to develop acute leukemia than African-Americans, Asians and Hispanics and tend to have a better prognosis than non-Caucasians. Age at diagnosis: children between 1– 10 years of age are most likely to develop ALL and to be cured of it. Cases in older patients are more likely to result from chromosomal abnormalities (e. g. the Philadelphia chromosome) that make treatment more difficult and prognoses poorer. White blood cell count at diagnosis of less than 50, 000/µl Cancer spread into the Central_nervous_system (brain or spinal cord) has worse outcomes. Morphological, immunological, and genetic subtypes Patient's response to initial treatment Genetic disorders such as Down's Syndrome

Cytogenetics, the study of characteristic large changes in the chromosomes of cancer cells, is an important predictor of outcome. [13] Some cytogenetic subtypes have a worse prognosis than others
(q 21; q 23) Poor")
Cytogenetic change Risk category Philadelphia chromosome Poor prognosis t(4; 11)(q 21; q 23) Poor prognosis t(8; 14)(q 24. 1; q 32) Poor prognosis Complex karyotype (more than four abnormalities) Poor prognosis Low hypodiploidy or near triploidy Poor prognosis High hyperdiploidy (specifically, trisomy 4, 10, 17) Good prognosis del(9 p) Good prognosis
4d36ba4410f4aa353cdcf7cebe1abcfa.ppt