

494ab5816dcca0c99f1c8b2d7eac519f.ppt
- Количество слайдов: 47
 Caratterizzazione funzionale di trascritti attraverso l'individuazione di elementi di controllo post-trascrizionale Graziano PESOLE Università di Milano 7 Marzo 2002 graziano. pesole@unimi. it
Caratterizzazione funzionale di trascritti attraverso l'individuazione di elementi di controllo post-trascrizionale Graziano PESOLE Università di Milano 7 Marzo 2002 graziano. pesole@unimi. it
 Microarray Experiment Result
Microarray Experiment Result
 Gene characterization problem Gene classification problem
Gene characterization problem Gene classification problem
 Gene characterization problem CDS 5’ m. RNA 3’ 3’ ESTs EST assembling • known gene • gene identification through database searching (e. g. BLASTX) • gene identification by aa pattern discovery (e. g. Pfam, etc. ) • gene identification by nt pattern discovery (e. g. UTRsite patterns)
Gene characterization problem CDS 5’ m. RNA 3’ 3’ ESTs EST assembling • known gene • gene identification through database searching (e. g. BLASTX) • gene identification by aa pattern discovery (e. g. Pfam, etc. ) • gene identification by nt pattern discovery (e. g. UTRsite patterns)
 Gene characterization problem CDS 5’ m. RNA 3’ protein CDS DNA Eventual alternatively spliced m. RNAs
Gene characterization problem CDS 5’ m. RNA 3’ protein CDS DNA Eventual alternatively spliced m. RNAs
 Gene characterization problem CDS 5’ 3’ EST m. RNA 3’UTR 3’ ?
Gene characterization problem CDS 5’ 3’ EST m. RNA 3’UTR 3’ ?
 post-transcriptional regulation of gene expression 5’UTR CDS 3’UTR SUBCELLULAR LOCALIZATION STABILITY m. RNA TRANSLATION
post-transcriptional regulation of gene expression 5’UTR CDS 3’UTR SUBCELLULAR LOCALIZATION STABILITY m. RNA TRANSLATION
 Biological role of m. RNA UTRs Function nuclear export polyadenylation status m. RNA cellular localization Control of m. RNA stability Control of m. RNA translation mediated by oligonucleotide patterns stem-loop structures
Biological role of m. RNA UTRs Function nuclear export polyadenylation status m. RNA cellular localization Control of m. RNA stability Control of m. RNA translation mediated by oligonucleotide patterns stem-loop structures
 ¨Novel patterns candidate") ¨Patterns known to play some regulatory activity (experimental assay, phylogenetic footprinting) ¨Novel patterns candidate of functional activity (Statistical analyses carried out on non-redundant databases)
¨Patterns known to play some regulatory activity (experimental assay, phylogenetic footprinting) ¨Novel patterns candidate of functional activity (Statistical analyses carried out on non-redundant databases)
") UTRdb - release 14. 0 (January 2001)
UTRdb - release 14. 0 (January 2001)
") Functional Elements annotation (UTRsite database)
Functional Elements annotation (UTRsite database)
 Consensus Oligonucleotides Consensus Secondary Structures
Consensus Oligonucleotides Consensus Secondary Structures
![Regular Expression Strings e. g. AAUAAA C-x(2, 4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3, 5)-H polyadenylation site C 2 H](https://present5.com/presentation/494ab5816dcca0c99f1c8b2d7eac519f/image-13.jpg "Regular Expression Strings e. g. AAUAAA C-x(2, 4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3, 5)-H polyadenylation site C 2 H") Regular Expression Strings e. g. AAUAAA C-x(2, 4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3, 5)-H polyadenylation site C 2 H 2 zinc-finger (deterministic description of the pattern : YES/NO) Consensus Matrices (probabilistic description of the pattern : threshold value)
Regular Expression Strings e. g. AAUAAA C-x(2, 4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3, 5)-H polyadenylation site C 2 H 2 zinc-finger (deterministic description of the pattern : YES/NO) Consensus Matrices (probabilistic description of the pattern : threshold value)
 present in the 3'untranslated region of mature lymphokine and cytokine m.") AU-rich elements (AREs) present in the 3'untranslated region of mature lymphokine and cytokine m. RNAs regulate m. RNA stability and translational efficiency. Based on their sequence features and functional properties AREs can be divided into three classes. Class II AREs direct asynchronous cytoplasmic deadenylation (processive kinetics)generating poly-A(-) m. RNAs. Among m. RNAs with class II ARE in their 3' UTR are GM-CSF, IL-2. The minimum number of AUUUA tandem motifs to activate processive degradation is 4 (of which at least 3 in tandem). Furthermore, an AU-rich region 20 -30 nt long immediately 5' to the cluster of AUUUA motifs can greatly enhance the degradation activity. CDS 5’UTR auuua An au-rich 3’UTR ARE II
AU-rich elements (AREs) present in the 3'untranslated region of mature lymphokine and cytokine m. RNAs regulate m. RNA stability and translational efficiency. Based on their sequence features and functional properties AREs can be divided into three classes. Class II AREs direct asynchronous cytoplasmic deadenylation (processive kinetics)generating poly-A(-) m. RNAs. Among m. RNAs with class II ARE in their 3' UTR are GM-CSF, IL-2. The minimum number of AUUUA tandem motifs to activate processive degradation is 4 (of which at least 3 in tandem). Furthermore, an AU-rich region 20 -30 nt long immediately 5' to the cluster of AUUUA motifs can greatly enhance the degradation activity. CDS 5’UTR auuua An au-rich 3’UTR ARE II
 cytoplasmic polyadenylylation Cytoplasmic polyadenylation is an evolutionarily conserved mechanism regulating translational activation of a set of quiescent maternal messenger RNAs (m. RNAs) during early development. Cytoplasmic poly(A) elongation occurs in a wide range of species, ranging from clam to mouse. The relevance of this process during late oogenesis and early embryogenesis has been shown by studies in the mouse, Xenopus, and Drosophila. In mouse and Xenopus, the only vertebrates so far examined, the critical regulatory sequences, referred to as Cytoplasmic Polyadenylation Elements (CPEs), are AU-rich and located in the 3' untranslated region (3'-UTR) near the canonical nuclear polyadenylation element (AAUAAA), which also is required for proper poly(A) addition. The CPE has the general structure of UUUUUUAU. However, the CPE is not identical in all m. RNAs and its position varies relative to AAUAAA (generally within 100 nucleotides). The minimal CPE capable to stimulate elongation of a poly(A) tail appears to be UUUUAU, and recent experiments show also the existence of substantial context and position effects on CPE function. CDS 5’UTR uuuuuuau aauaaa 3’UTR CPE An
cytoplasmic polyadenylylation Cytoplasmic polyadenylation is an evolutionarily conserved mechanism regulating translational activation of a set of quiescent maternal messenger RNAs (m. RNAs) during early development. Cytoplasmic poly(A) elongation occurs in a wide range of species, ranging from clam to mouse. The relevance of this process during late oogenesis and early embryogenesis has been shown by studies in the mouse, Xenopus, and Drosophila. In mouse and Xenopus, the only vertebrates so far examined, the critical regulatory sequences, referred to as Cytoplasmic Polyadenylation Elements (CPEs), are AU-rich and located in the 3' untranslated region (3'-UTR) near the canonical nuclear polyadenylation element (AAUAAA), which also is required for proper poly(A) addition. The CPE has the general structure of UUUUUUAU. However, the CPE is not identical in all m. RNAs and its position varies relative to AAUAAA (generally within 100 nucleotides). The minimal CPE capable to stimulate elongation of a poly(A) tail appears to be UUUUAU, and recent experiments show also the existence of substantial context and position effects on CPE function. CDS 5’UTR uuuuuuau aauaaa 3’UTR CPE An
 m. RNA patterns usually located in 5’ or 3’ UTRs that are able to fold into specific secondary structures able to be recognized by specific RNA binding proteins
m. RNA patterns usually located in 5’ or 3’ UTRs that are able to fold into specific secondary structures able to be recognized by specific RNA binding proteins
 Histone 3’UTR m. RNA element Metazoan histone 3'-UTR m. RNAs, lacking a poly. A tail, contain a highly conserved stem-loop structure with a six base stem and a four base loop. This stemloop structure plays a different role in the nucleus and in the cytoplasm. In the nucleus, it is involved in pre-m. RNA processing and nucleocytoplasmic transport, whereas in the cytoplasm it enhances translation efficiency and regulates histone m. RNA stability. The trans-acting factor which interacts with the 3'-UTR hairpin structure of histone m. RNAs is a 31 k. Da stem-loop binding protein in mammals (SLBP) present both in nuclei and polyribosomes. In mammals in addition to SLBP histone m. RNA processing requires at least one additional factor: the U 7 sn. RNP, which binds a purine-rich element 10 -20 nt downstream of the stem-loop sequence (Histone Downstream Element, HDE). The histone 3'-UTR hairpin structure is peculiar in that the bases of the stem are conserved unlike most functional hairpin motifs where conserved bases are found in single stranded loop regions only. The sequence of the stem an flanking sequences are critical for binding of the SLBP.
Histone 3’UTR m. RNA element Metazoan histone 3'-UTR m. RNAs, lacking a poly. A tail, contain a highly conserved stem-loop structure with a six base stem and a four base loop. This stemloop structure plays a different role in the nucleus and in the cytoplasm. In the nucleus, it is involved in pre-m. RNA processing and nucleocytoplasmic transport, whereas in the cytoplasm it enhances translation efficiency and regulates histone m. RNA stability. The trans-acting factor which interacts with the 3'-UTR hairpin structure of histone m. RNAs is a 31 k. Da stem-loop binding protein in mammals (SLBP) present both in nuclei and polyribosomes. In mammals in addition to SLBP histone m. RNA processing requires at least one additional factor: the U 7 sn. RNP, which binds a purine-rich element 10 -20 nt downstream of the stem-loop sequence (Histone Downstream Element, HDE). The histone 3'-UTR hairpin structure is peculiar in that the bases of the stem are conserved unlike most functional hairpin motifs where conserved bases are found in single stranded loop regions only. The sequence of the stem an flanking sequences are critical for binding of the SLBP.
 Specific incorporation of selenocysteine in selenoproteins is directed by") SEleno. Cysteine Insertion Sequence (SECIS) Specific incorporation of selenocysteine in selenoproteins is directed by UGA codons residing within the coding sequence of the corresponding m. RNAs. Translation of UGA, usually a termination codon, as selenocysteine requires a conserved stem-loop structure called "SEleno. Cysteine Insertion Sequence" (SECIS) lying in the 3'UTR region of selenoprotein m. RNAs. The consensus structure of SECIS element determined by comparative analysis of several selenoprotein m. RNAs as well as on both RNase and chemical probing. UGA SECIS
SEleno. Cysteine Insertion Sequence (SECIS) Specific incorporation of selenocysteine in selenoproteins is directed by UGA codons residing within the coding sequence of the corresponding m. RNAs. Translation of UGA, usually a termination codon, as selenocysteine requires a conserved stem-loop structure called "SEleno. Cysteine Insertion Sequence" (SECIS) lying in the 3'UTR region of selenoprotein m. RNAs. The consensus structure of SECIS element determined by comparative analysis of several selenoprotein m. RNAs as well as on both RNase and chemical probing. UGA SECIS
 nanos TCE Localization of nanos RNA to the posterior pole of the embryo is essential for translation of Nanos protein. In situ hybridization to nanos RNA (top), antibody staining with anti-Nanos antibody (bottom). The 3'UTR of drosophila nanos m. RNA contains the essential signals for generating the nanos gradient emanating from the posterior pole of drosophila embryos. The polarized distribution of nanos is generated by the localization dependent translation of nanos m. RNA in the pole plasm. The translation control element (TCE) consists of a a 90 -nt region located in the 3'UTR of nanos m. RNA which is able to fold into a bipartite secondary structure that is recognized by Smaug repressor and at least one additional factor. A stem-loop bearing the CUGGC pentamer in the loop is required for Smaug: TCE interaction whereas both sequence and structure of another stem-loop is critical for TCE function. Translation activation is mediated by the interaction of localization factors with a 540 -nt sequence regions overlapping the TCE structural motif.
nanos TCE Localization of nanos RNA to the posterior pole of the embryo is essential for translation of Nanos protein. In situ hybridization to nanos RNA (top), antibody staining with anti-Nanos antibody (bottom). The 3'UTR of drosophila nanos m. RNA contains the essential signals for generating the nanos gradient emanating from the posterior pole of drosophila embryos. The polarized distribution of nanos is generated by the localization dependent translation of nanos m. RNA in the pole plasm. The translation control element (TCE) consists of a a 90 -nt region located in the 3'UTR of nanos m. RNA which is able to fold into a bipartite secondary structure that is recognized by Smaug repressor and at least one additional factor. A stem-loop bearing the CUGGC pentamer in the loop is required for Smaug: TCE interaction whereas both sequence and structure of another stem-loop is critical for TCE function. Translation activation is mediated by the interaction of localization factors with a 540 -nt sequence regions overlapping the TCE structural motif.

 Pat. Search is a pattern matcher which searches protein or nucleotide (DNA, RNA, t. RNA, and so on) sequences in order to find instances of a pattern which you can give as input. It is able to find, in a given sequence, kinds of loop structures that characterize t. RNAs, r. RNAs (hairpin loop, stem loop with bulges or internal loops) and/or any kind of pattern in DNA and protein sequences. Pat. Search also allows to use non-standard pairings for reverse matching and tolerate some numbers of mismatches and bulges.
Pat. Search is a pattern matcher which searches protein or nucleotide (DNA, RNA, t. RNA, and so on) sequences in order to find instances of a pattern which you can give as input. It is able to find, in a given sequence, kinds of loop structures that characterize t. RNAs, r. RNAs (hairpin loop, stem loop with bulges or internal loops) and/or any kind of pattern in DNA and protein sequences. Pat. Search also allows to use non-standard pairings for reverse matching and tolerate some numbers of mismatches and bulges.
 A pattern is a sequence of simple pattern units. A simple pattern unit is either a named units pattern unit, a complementation rule pattern unit or a basic pattern unit. In pattern definition, upper and lower case can be used interchangeably. Named Pattern Unit Complementation rule pattern unit Name=Basic. Pattern Name=Complements p 1=4. . . 7 or p 1=ACGTAGT r 1={au, ua, gc, cg} Basic. Pattern: 1. String of characters to be matched (including standard ambiguity codes for nucleotides and character X for proteins), optionally followed by a match qualifier of the form [Mismatches, Deletions, Insertions] Example: TATAA[1, 0, 0] match TATAA, allowing 1 mismatch 2. Range pattern unit of the form “Min. . . Max” Example: 0. . . 5 match 1 to 5 characters or none 3. Complement pattern unit that is used to match the reverse complement of a named pattern unit, previously defined. Example: r 1~p 2 match the reverse complement of p 2 using rules r 1 4. Length-limit pattern unit puts a upper or lower bound on the sum of the lengths matched by previous named pattern units. Example: length(p 1+p 2+p 3) < 9 5. Weight pattern unit is used for matching against nucleotide sequences by using a weight matrix (Log-odds scoring method or matrix similarity method).
A pattern is a sequence of simple pattern units. A simple pattern unit is either a named units pattern unit, a complementation rule pattern unit or a basic pattern unit. In pattern definition, upper and lower case can be used interchangeably. Named Pattern Unit Complementation rule pattern unit Name=Basic. Pattern Name=Complements p 1=4. . . 7 or p 1=ACGTAGT r 1={au, ua, gc, cg} Basic. Pattern: 1. String of characters to be matched (including standard ambiguity codes for nucleotides and character X for proteins), optionally followed by a match qualifier of the form [Mismatches, Deletions, Insertions] Example: TATAA[1, 0, 0] match TATAA, allowing 1 mismatch 2. Range pattern unit of the form “Min. . . Max” Example: 0. . . 5 match 1 to 5 characters or none 3. Complement pattern unit that is used to match the reverse complement of a named pattern unit, previously defined. Example: r 1~p 2 match the reverse complement of p 2 using rules r 1 4. Length-limit pattern unit puts a upper or lower bound on the sum of the lengths matched by previous named pattern units. Example: length(p 1+p 2+p 3) < 9 5. Weight pattern unit is used for matching against nucleotide sequences by using a weight matrix (Log-odds scoring method or matrix similarity method).
 Histone 3’UTR m. RNA element Pat. Search pattern: r 1={au, ua, gc, cg, gu, ug} n mmm p 1=ggyyy u hhuh a r 1~p 1 mm 0… 3 (m=a/c; y=c/u; h=not g)
Histone 3’UTR m. RNA element Pat. Search pattern: r 1={au, ua, gc, cg, gu, ug} n mmm p 1=ggyyy u hhuh a r 1~p 1 mm 0… 3 (m=a/c; y=c/u; h=not g)
 AUG Pat. Search syntax: r 1={au, ua, cg, gc,") Internal Ribosome Entry Site (IRES) AUG Pat. Search syntax: r 1={au, ua, cg, gc, gu, ug} p 1=5. . . 6 0. . . 6 p 2=5. . . 6 p 3=0. . . 2 p 4=5. . . 8 p 5=3. . . 8 r 1~p 4[1, 0, 0] p 6=5. . . 8 p 7=3. . . 5 r 1~p 6[1, 0, 0] p 8=0. . . 5 r 1~p 2[1, 0, 0] 0. . . 6 r 1~p 1[1, 0, 0] 2. . . 5 p 9=5. . . 6 p 10=3. . . 8 r 1~p 9[1, 0, 0] 3. . . 10 $
Internal Ribosome Entry Site (IRES) AUG Pat. Search syntax: r 1={au, ua, cg, gc, gu, ug} p 1=5. . . 6 0. . . 6 p 2=5. . . 6 p 3=0. . . 2 p 4=5. . . 8 p 5=3. . . 8 r 1~p 4[1, 0, 0] p 6=5. . . 8 p 7=3. . . 5 r 1~p 6[1, 0, 0] p 8=0. . . 5 r 1~p 2[1, 0, 0] 0. . . 6 r 1~p 1[1, 0, 0] 2. . . 5 p 9=5. . . 6 p 10=3. . . 8 r 1~p 9[1, 0, 0] 3. . . 10 $
 http: //bigarea. ba. cnr. it: 8000/Emb. IT/Patsearch. html
http: //bigarea. ba. cnr. it: 8000/Emb. IT/Patsearch. html
 Search of the Histone 3’UTR motif in EST sequences EM_EST 1: AA 545838 EM_EST 1: AA 545875 EM_EST 1: AA 545884 EM_EST 1: AA 563524 EM_EST 1: AA 610040 : [420, 442] : [369, 391] : [419, 441] : [422, 444] : [403, 425] : C : C : C CAAC CAAA GGCCC GGCTC T T T CTTT TTTC A A A GGGCC GAGCC AC AC AC EM_EST 2: AA 701695 : [155, 177] : C CAAC GGCCC T CTTT A GGGCC AC Entry name Database Division Position Matched pattern Given that only about ONE random hit of this pattern is expected in 100 Mb we can reliably assign the matched ESTs to m. RNAs coding for histone-related proteins
Search of the Histone 3’UTR motif in EST sequences EM_EST 1: AA 545838 EM_EST 1: AA 545875 EM_EST 1: AA 545884 EM_EST 1: AA 563524 EM_EST 1: AA 610040 : [420, 442] : [369, 391] : [419, 441] : [422, 444] : [403, 425] : C : C : C CAAC CAAA GGCCC GGCTC T T T CTTT TTTC A A A GGGCC GAGCC AC AC AC EM_EST 2: AA 701695 : [155, 177] : C CAAC GGCCC T CTTT A GGGCC AC Entry name Database Division Position Matched pattern Given that only about ONE random hit of this pattern is expected in 100 Mb we can reliably assign the matched ESTs to m. RNAs coding for histone-related proteins
 ¨Novel patterns candidate") ¨Patterns known to play some regulatory activity (experimental assay, phylogenetic footprinting) ¨Novel patterns candidate of functional activity (Statistical analyses carried out on non-redundant databases)
¨Patterns known to play some regulatory activity (experimental assay, phylogenetic footprinting) ¨Novel patterns candidate of functional activity (Statistical analyses carried out on non-redundant databases)
 SOM clustering (b) Hierarchical clustering") Monitoring gene expression using DNA microarrays (a) SOM clustering (b) Hierarchical clustering
Monitoring gene expression using DNA microarrays (a) SOM clustering (b) Hierarchical clustering
 If you have a group of genes with a similar expression profile (e. g. those activated at the same time in cell-cycle) a natural assumption is that this profile is, at least partly, caused by and reflected in a similar structure of regions involved in transcription regulation.
If you have a group of genes with a similar expression profile (e. g. those activated at the same time in cell-cycle) a natural assumption is that this profile is, at least partly, caused by and reflected in a similar structure of regions involved in transcription regulation.
 common") Research has thus focused on the detection of motifs (possibly representing TF-bindiing sites) common to the promoter sequences of putatively coregulated genes.
Research has thus focused on the detection of motifs (possibly representing TF-bindiing sites) common to the promoter sequences of putatively coregulated genes.
 Significant Pattern Random Pattern
Significant Pattern Random Pattern
 v Occurrence v Position v Information Content
v Occurrence v Position v Information Content
 v Occurrence The number of sequences containing a given pattern should be significantly greater than expected (e. g. Word. UP algorithm). PATTERNS OBSERVED EXPECTED CHI-SQUARE AATAAA 1345 414. 00035 2093. 62225 AAATAA 834 414. 00035 426. 08588 ATAAAG 578 258. 12928 396. 37997 ATAAAA 744 414. 00035 263. 04270 CCCCCC 273 654. 61047 222. 46291 ATAAAT 584 321. 22291 214. 96537 GAAATA 443 239. 14498 173. 77269 TAAATA 496 285. 44362 155. 31610 TGTATTT 243 103. 18333 189. 45602 TGTATAT 154 56. 34083 169. 27891 ATATTTA 221 95. 25445 165. 99689 TTTATAT 218 103. 87432 125. 38875 TGTACAT 130 50. 48650 125. 22942 ATATATA 136 59. 08119 100. 14193 GCGGCCGC 38 5. 41527 196. 06842 ATATATTT 100 31. 42024 149. 68643 GGGTGGGG 92 31. 82251 113. 79774 TTTAAAAA 211 103. 89444 110. 41593 TACATTTT 92 33. 40711 102. 76638 TATTT 94 16. 60269 360. 80574 TTTTTAAAA 139 40. 25077 242. 26645
v Occurrence The number of sequences containing a given pattern should be significantly greater than expected (e. g. Word. UP algorithm). PATTERNS OBSERVED EXPECTED CHI-SQUARE AATAAA 1345 414. 00035 2093. 62225 AAATAA 834 414. 00035 426. 08588 ATAAAG 578 258. 12928 396. 37997 ATAAAA 744 414. 00035 263. 04270 CCCCCC 273 654. 61047 222. 46291 ATAAAT 584 321. 22291 214. 96537 GAAATA 443 239. 14498 173. 77269 TAAATA 496 285. 44362 155. 31610 TGTATTT 243 103. 18333 189. 45602 TGTATAT 154 56. 34083 169. 27891 ATATTTA 221 95. 25445 165. 99689 TTTATAT 218 103. 87432 125. 38875 TGTACAT 130 50. 48650 125. 22942 ATATATA 136 59. 08119 100. 14193 GCGGCCGC 38 5. 41527 196. 06842 ATATATTT 100 31. 42024 149. 68643 GGGTGGGG 92 31. 82251 113. 79774 TTTAAAAA 211 103. 89444 110. 41593 TACATTTT 92 33. 40711 102. 76638 TATTT 94 16. 60269 360. 80574 TTTTTAAAA 139 40. 25077 242. 26645
 v Position Functional patterns are generally in conserved positions (e. g. within a specific distance range from the transcription start site).
v Position Functional patterns are generally in conserved positions (e. g. within a specific distance range from the transcription start site).
 v Information Content The functional constraints on each specific position of the pattern are variable from some sites absolutely conserved (Shannon’s information content Ci ranging between 0 and 1).
v Information Content The functional constraints on each specific position of the pattern are variable from some sites absolutely conserved (Shannon’s information content Ci ranging between 0 and 1).
 Ten random sequences 1 kb long, each containing the same 15 -mer motif (up to 2 mismatches allowed).
Ten random sequences 1 kb long, each containing the same 15 -mer motif (up to 2 mismatches allowed).
 Two different approaches can be used to extract functional motifs from regulatory regions of coregulated genes: v Alignment methods v Enumeration or exaustive methods
Two different approaches can be used to extract functional motifs from regulatory regions of coregulated genes: v Alignment methods v Enumeration or exaustive methods
 Try to identify unknown signals by a significant local multiple alignment of all sequences v Gibbs sampling v MEME system (expectation maximization) These methods use a statistical approach that considers unknown the start position of the motifs in the sequences and perform a local optimization to determine which positions deliver the “optimal” motifs.
Try to identify unknown signals by a significant local multiple alignment of all sequences v Gibbs sampling v MEME system (expectation maximization) These methods use a statistical approach that considers unknown the start position of the motifs in the sequences and perform a local optimization to determine which positions deliver the “optimal” motifs.
 http: //bayesweb. wadsworth. org/gibbs. html Two approaches Site sampling : each sequence is assumed to contain one motif element for each motif type Motif sampling : each sequence is assumed to contain one motif element for each motif type
http: //bayesweb. wadsworth. org/gibbs. html Two approaches Site sampling : each sequence is assumed to contain one motif element for each motif type Motif sampling : each sequence is assumed to contain one motif element for each motif type
 http: //meme. sdsc. edu/meme/website/
http: //meme. sdsc. edu/meme/website/
 v YEBIS") v Word. UP (http: //bio-www. ba. cnr. it: 8000/Bio. WWW/wordup. GCG. html) v YEBIS (http: //www-scc. jst. go. jp: 8080/sankichi/Motif. Extraction/) v Pratt (http: //www. ii. uib. no/~inge/Pratt. html/) v Verbumculus (http: //www. cs. purdue. edu/homes/stelo/Verbumculus/) v RSA tools (http: //copan. cifn. unam. mx/~jvanheld/rsa-tools/) (test dataset: 20 sequences 100 nt long with a 8 -mer shared by 75% of them)
v Word. UP (http: //bio-www. ba. cnr. it: 8000/Bio. WWW/wordup. GCG. html) v YEBIS (http: //www-scc. jst. go. jp: 8080/sankichi/Motif. Extraction/) v Pratt (http: //www. ii. uib. no/~inge/Pratt. html/) v Verbumculus (http: //www. cs. purdue. edu/homes/stelo/Verbumculus/) v RSA tools (http: //copan. cifn. unam. mx/~jvanheld/rsa-tools/) (test dataset: 20 sequences 100 nt long with a 8 -mer shared by 75% of them)
 ---------------------------------------OCCURRENCES ---------------------------------------WORDS OBSERVED EXPECTED CHI-SQUARE ---------------------------------------GGATCAA ATCAAA AGGATC TCAAAG GTGGAT GCAAGC GTTTGG CAAAGT 15 15 15 7 6 7 4 5 4 4 0. 58 0. 63 0. 65 0. 57 0. 48 0. 66 0. 45 0. 69 0. 49 0. 56 361. 51 325. 96 316. 96 72. 70 63. 10 60. 58 28. 05 27. 00 25. 09 21. 22 ---------------------------------------OCCURRENCES ---------------------------------------WORDS OBSERVED EXPECTED CHI-SQUARE ---------------------------------------AGGATC GCAAGC GTTTGG TCAAAGT GTGGATCAAA 7 5 4 4 3 15 0. 57 0. 69 0. 49 0. 16 0. 11 0. 04 72. 70 27. 00 25. 09 92. 97 74. 04 5415. 19
---------------------------------------OCCURRENCES ---------------------------------------WORDS OBSERVED EXPECTED CHI-SQUARE ---------------------------------------GGATCAA ATCAAA AGGATC TCAAAG GTGGAT GCAAGC GTTTGG CAAAGT 15 15 15 7 6 7 4 5 4 4 0. 58 0. 63 0. 65 0. 57 0. 48 0. 66 0. 45 0. 69 0. 49 0. 56 361. 51 325. 96 316. 96 72. 70 63. 10 60. 58 28. 05 27. 00 25. 09 21. 22 ---------------------------------------OCCURRENCES ---------------------------------------WORDS OBSERVED EXPECTED CHI-SQUARE ---------------------------------------AGGATC GCAAGC GTTTGG TCAAAGT GTGGATCAAA 7 5 4 4 3 15 0. 57 0. 69 0. 49 0. 16 0. 11 0. 04 72. 70 27. 00 25. 09 92. 97 74. 04 5415. 19
 Pratt is able to find patterns of a quite general form. For example, Pratt found in less than 30 seconds the pattern C-x(2, 4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3, 5)-H conserved in a set of 285 zinc finger protein sequenes. The sequences were given to Pratt unaligned, and no information was given about the pattern. Pratt will not always find interesting patterns this quickly, but it is a powerful tool that allows you to discovery conserved patterns when they exist. Best patterns with alignments: fitness hits(seqs) Pattern A 1: 12. 0000 11( 10) C-x-T-x(2)-C-C-T-C rand_1: 1927: cgagg Cc. Ttt. CCTC ccagg rand_2: 399407: gatga Ct. Tcg. CCTC gccct rand_3: 424432: cgaag Cc. Tct. CCTC cccag rand_3: 456464: tgagg Cg. Tgc. CCTC ctcac rand_4: 689697: cgagg Cc. Ttc. CCTC ccacg rand_5: 100108: ggaag Cc. Tct. CCTC ccgtc rand_6: 187195: gaaac Ct. Tcg. CCTC agttt rand_7: 1002 - 1010: cgaag Cc. Tct. CCTC ccttt rand_8: 233241: cctgg Ct. Tac. CCTC gattc rand_9: 715: gagaa Ca. Taa. CCTC cggcc rand_10: 980988: ttgcc Ca. Ttc. CCTC gcagg
Pratt is able to find patterns of a quite general form. For example, Pratt found in less than 30 seconds the pattern C-x(2, 4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3, 5)-H conserved in a set of 285 zinc finger protein sequenes. The sequences were given to Pratt unaligned, and no information was given about the pattern. Pratt will not always find interesting patterns this quickly, but it is a powerful tool that allows you to discovery conserved patterns when they exist. Best patterns with alignments: fitness hits(seqs) Pattern A 1: 12. 0000 11( 10) C-x-T-x(2)-C-C-T-C rand_1: 1927: cgagg Cc. Ttt. CCTC ccagg rand_2: 399407: gatga Ct. Tcg. CCTC gccct rand_3: 424432: cgaag Cc. Tct. CCTC cccag rand_3: 456464: tgagg Cg. Tgc. CCTC ctcac rand_4: 689697: cgagg Cc. Ttc. CCTC ccacg rand_5: 100108: ggaag Cc. Tct. CCTC ccgtc rand_6: 187195: gaaac Ct. Tcg. CCTC agttt rand_7: 1002 - 1010: cgaag Cc. Tct. CCTC ccttt rand_8: 233241: cctgg Ct. Tac. CCTC gattc rand_9: 715: gagaa Ca. Taa. CCTC cggcc rand_10: 980988: ttgcc Ca. Ttc. CCTC gcagg
 Suffix Tree
Suffix Tree
 v The motif size may be unknown v The motif may not be well conserved between sequences
v The motif size may be unknown v The motif may not be well conserved between sequences
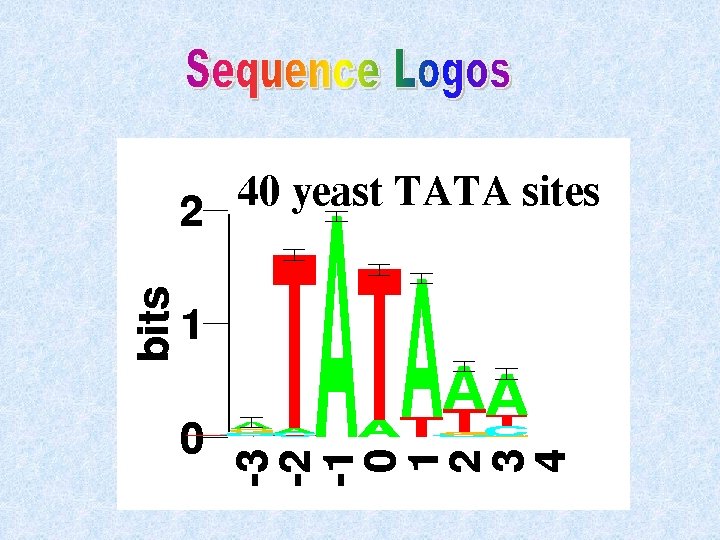
 v The motif size is unknown v The motif may not be well conserved between sequences v The sequences used for the search does not necessarily represent a homogeneous set (expression clustering algorithms artefacts) v The assessment of statistical significance (suitable background model) v Motifs allowing gaps or variable spacers
v The motif size is unknown v The motif may not be well conserved between sequences v The sequences used for the search does not necessarily represent a homogeneous set (expression clustering algorithms artefacts) v The assessment of statistical significance (suitable background model) v Motifs allowing gaps or variable spacers