болезни геномного импринтинга.ppt
- Количество слайдов: 20



Болезни геномного импринтинга

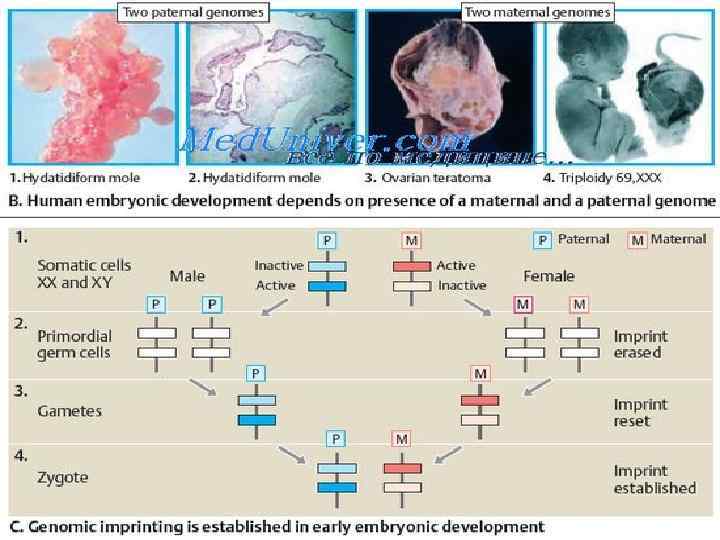
Геномный импринтинг Среди наследственных болезней, не наследующихся по классическим законам Менделя, в последние годы выделены болезни, связанные с вновь открытым генетическим явлением - геномным импринтингом.


Синдром Прадера — Вилли Синдром Пра дера — Ви лли — редкая генетическая аномалия. При синдроме Прадера — Вилли отсутствуют примерно 7 генов из 15 хромосомы. Кариотип 46 XX или ХУ, 15 q-11 -q 12. Синдром впервые описали в 1956 г. Андреа Прадер, Хайнрих Вилли, Алексис Лабхарт, Эндрю Зиглер и Гвидо Фанцони в Швейцарии. Частота встречаемости — 1 : 12 000 — 15 000 живорождённых младенцев. При синдроме Прадера-Вилли страдает отцовская хромосома

Особенности до рождения: низкая подвижность плода; n часто — неправильное положение плода; n ожирение; склонность к перееданию; n пониженный мышечный тонус (гипотонус); пониженная координация движений; n маленькие кисти и стопы, низкий рост; n повышенная сонливость; n страбизм (косоглазие); n сколиоз (искривление позвоночника); n пониженная плотность костей; n сниженная функция половых желёз (гипогонадизм); в результате, как правило, бесплодие; n речевая задержка, задержка психического развития; отставание в освоении навыков общей и мелкой моторики. n более позднее половое созревание. Внешние признаки: у взрослых выражена переносица; лоб высокий и узкий; глаза, как правило, миндалевидные; губы узкие. n
.")
Диагностика Синдром диагностируется путём генетического анализа, рекомендуемого для новорождённых с пониженным мышечным тонусом (гипотонусом). Иногда вместо диагноза «синдром Прадера — Вилли» врачи ошибочно ставят диагноз «синдром Дауна» (поскольку синдром Дауна встречается намного чаще).

Перспективы развития У большинства людей с синдромом Прадера — Вилли наблюдается задержка психического и речевого развития. Согласно исследованиям Керфс и Фрим (1992), n 5 % обследованных продемонстрировали средний уровень коэффициента интеллекта (более 85 баллов по шкале IQ); n 27 % — уровень на грани среднего (70 -85 баллов); n 34 % — уровень слабого отставания (50 -70 баллов); n 27 % — уровень среднего отставания (35 -70 баллов); n 5 % — сильное отставание (20 -35 баллов); n менее 1 % — значительное отставание.

Синдром Ангельмана — генетическая аномалия. Для него характерны задержка психического развития, нарушения сна, припадки, хаотические движения (особенно рук), частый смех или улыбки. Кариотип 46 XX или XY, 15 р−. Обычно синдром вызывается спонтанным хромосомным дефектом, когда отсутствует большая смежная область из 3— 4 миллионов пар оснований ДНК в области q 11—q 13 15 -й хромосомы. Согласно результатам многих независимых исследований, причиной возникновения синдрома Ангельмана может являться мутация в гене UBE 3 A. Продукт этого гена — ферментный компонент сложной системы деградации белков

Синдром назван по имени британского педиатра Гарри Ангельмана, впервые описавшего в 1965 г. Частота встречаемости, по разным данным, — 1 : 10 000— 20 000 живорожденных младенцев. Однако, согласно данным Центра развития человека и отклонений в развитии (университет Вашингтона, США), можно предполагать, что доля людей с синдромом Ангельмана в действительности намного больше статистической

Особенности n n n n n В 75 % проблемы с питанием, особенно с грудным вскармливанием, такие младенцы плохо набирают вес; задержка в развитии навыков общей моторики (умение сидеть, ходить); задержка речевого развития, неразвитая речь (у всех детей); дети больше понимают, чем могут сказать или выразить; дефицит внимания и гиперактивность; сложности с обучением; эпилепсия (80 % случаев), нарушения выявляются также при электроэнцефалографии; считается, что у детей с синдромом Ангельмана наблюдается вторичная (симптоматическая) эпилепсия; необычные движения (мелкий тремор, хаотические движения конечностей); частый смех без повода; ходьба на негнущихся ногах — из-за этой особенности детей с этим синдромом иногда сравнивали с марионетками; размер головы меньше среднего, нередко с уплощением затылка; иногда характерные черты лица — широкий рот, редко расположенные зубы, выдающийся вперед подбородок, высунутый наружу язык; нарушения сна; страбизм (косоглазие) в 40 % случаев; сколиоз (искривление позвоночника) в 10 % случаев; повышенная чувствительность к высокой температуре; чувствуют себя комфортнее в воде.
, рекомендуемого для новорожденных")
Диагностика n n n Синдром диагностируется путем генетического анализа (15 хромосома), рекомендуемого для новорожденных с пониженным мышечным тонусом (гипотонусом), отставанием в развитии общей моторики и в развитии речи. Родители и врачи должны обратить внимание на случаи мелкого тремора, хаотические, порывистые движения конечностей, походку с негнущимися ногами; в ряде случаев специфическое выражение лица, слишком частый смех. Возможные методы анализа: процесс флуоресцентной гибридизации in situ, метилирование ДНК в области 15 q 11— q 13, анализ мутации импринтингового центра, анализ прямой мутации гена UBE 3 A. Существует небольшая группа людей, у которых результаты всех вышеописанных анализов в норме, однако наблюдаются все внешние проявления синдрома Ангельмана.

,")
Синдром Видемана — Беквита Достаточно хорошо изучен в плане импринтинга также синдром Беквита—Видемана (СБВ), имеющий следующие основные признаки: макросомию, макроглоссию, пупочную грыжу, повышенную предрасположенность к опухолям. При нем обнаруживают структурные и функциональные аномалии критического района короткого плеча хромосомы 11. Гены-кандидаты для синдрома Беквита—Видемана (CDKN 1 C и IGF 2) расположены в этом регионе (11 p 15. 5), где находится кластер импринтированных генов.

Среди семейных случаев синдрома Беквита—Видемана почти в 40 % выявляют мутации гена CDKN 1 C, а в спорадических — не более 5 %. В гене, экспрессирующемся с отцовской хромосомы, мутации не выявляются, но у больных с этим синдромом найдена диаллельная экспрессия гена, т. е. ген начинает экспрессироваться с материнской хромосомы, либо диаллельная экспрессия является результатом отцовской ОРД.

Такое явление называют потерей импринтинга, его обнаруживают у 20 % больных синдромом Беквита— Видемана. Кроме того, многие другие гены из кластера генов, расположенных в этом регионе, проявляют эффект импринтинга, который может приводить к заболеванию. Суммарно структурную и функциональную патологию при синдроме Беквита— Видемана можно определить в 85— 95 % случаев.
 — комплекс наследственных аномалий (предполагается")
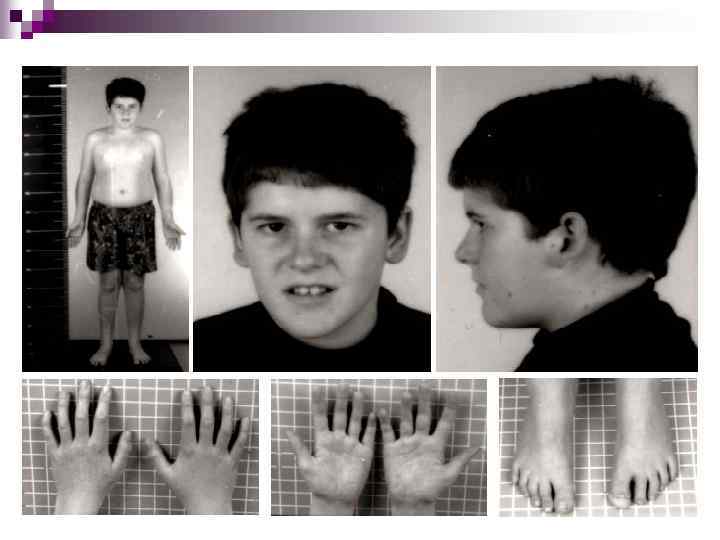
Синдром Рассела-Сильвера Синдро м Рассе ла-Си львера (карликовость Рассела-Сильвера) — комплекс наследственных аномалий (предполагается аутосомно-рецессивный тип наследования) врождённый карликовый рост в результате нарушений эмбрионального развития: малая масса тела при рождении, низкий рост, задержка общего развития, треугольное лицо, опущенные вниз уголки рта, укороченные и согнутые пальцы рук, синдактилия.

Впервые заболевание описано в середине прошлого века. Частота в популяции 1: 30 000. Встречается в большинстве случаев спорадически, однако описаны и единичные родословные с данной патологией. Тип наследования не установлен.
болезни геномного импринтинга.ppt