f2d95b5c2151e206cc4f5c773c232fa1.ppt
- Количество слайдов: 50



Bioinformatics Tools Stuart M. Brown, Ph. D Dept of Cell Biology NYU School of Medicine

Bioinformatics Tools Stuart M. Brown, Ph. D Dept of Cell Biology NYU School of Medicine

Overview This lecture will summarize a huge amount of bioinformatics material that is usually presented as a full 12 week course. – Data management and analysis of sequences from the HGP – A quick look at Gen. Bank and ENTREZ. – Gene finding and translation – Similarity searching and alignment (BLAST) – Protein structure and function

Data Management and Analysis • The Human Genome Project has generated huge quantities of DNA sequence data. • This data will lead to many medial advances. • But a great deal of analysis and research will be needed.

Access to the Data • Organize the genome data & provide access for scientists • Use the Internet • The data is public, so anyone can access it.

Gen. Bank • All Genome Project data is stored in a database called Gen. Bank managed by the National Center for Biotechnology Information (NCBI) • The NCBI is a branch of the National Library of Medicine, which is part of the NIH (National Institutes of Health). http: //ncbi. nlm. nih. gov
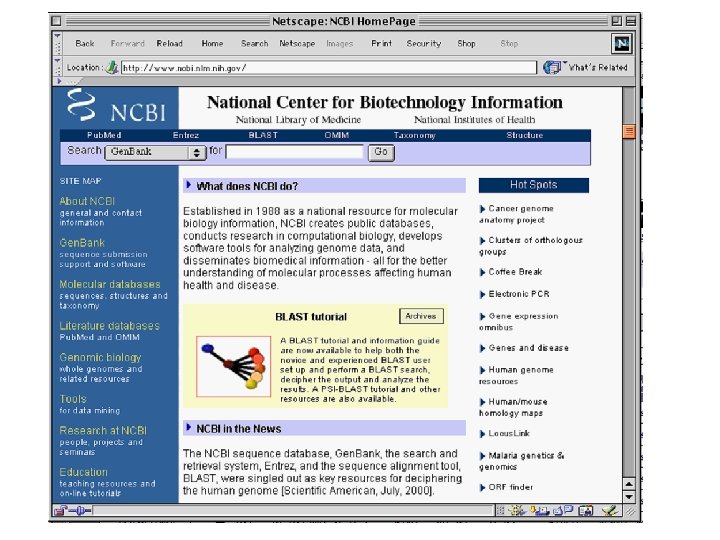

Gen. Bank Sections In addition to DNA sequences of genes Gen. Bank has a number of other sections including: • Protein sequences (translated from DNA) • Short RNA fragments (ESTs) • Cancer Genome Anatomy Project (CGAP) gene expression profiles of normal, pre-cancer, and cancer cells from a wide variety of tissue types • Single Nucleotide Polymorphisms (SNPs) which represent genetic variations in the human population • Online Mendelian Inheritance in Man (OMIM) a database of human genetic disorders

Finding Genes • Gen. Bank contains approximately 13 billion bases in 12 million sequence records (as of August 2001). • These billions of G, A, T, and C letters would be almost useless without descriptions of what genes they contain, the organisms they come from, etc. • All of this information is contained in the "annotation" part of each sequence record.


Entrez is a Tool for Finding Sequences • NCBI has created a Web-based tool called Entrez for finding sequences in Gen. Bank. • Each sequence in Gen. Bank has a unique “accession number”. • Entrez can also search for keywords such as gene names, protein names, and the names of orgainisms or biological functions
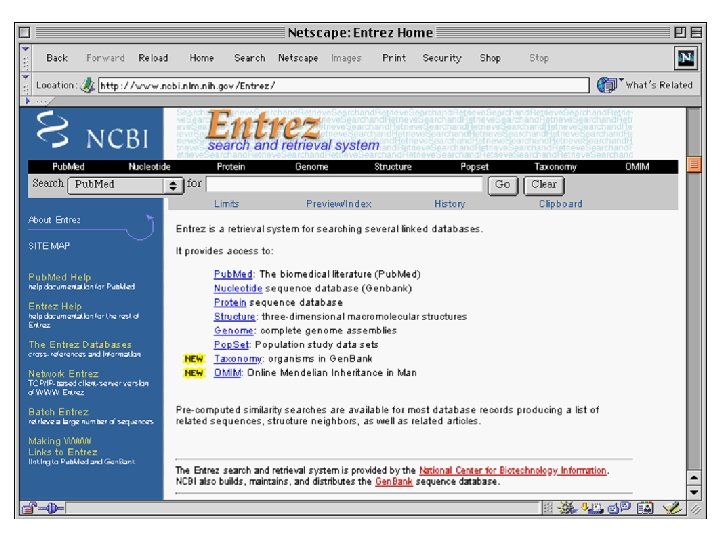

Entrez has links to Medline • Entrez is much more than just a tool for finding sequences by keywords. • It contains links to Pub. Med/Medline • Entrez also contains all known protein sequences and 3 -D protein structures.
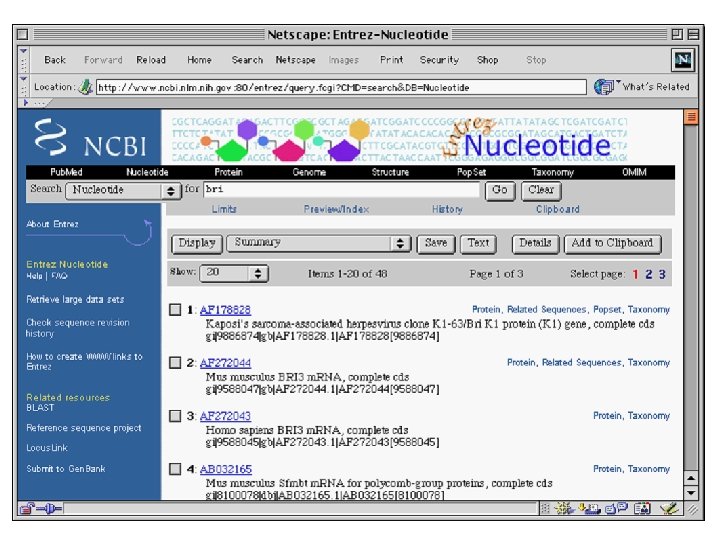

Entrez is Internally Cross-linked • DNA and protein sequences are linked to other similar sequences • Medline citations are linked to other citations that contain similar keywords • 3 -D structures are linked to similar structures
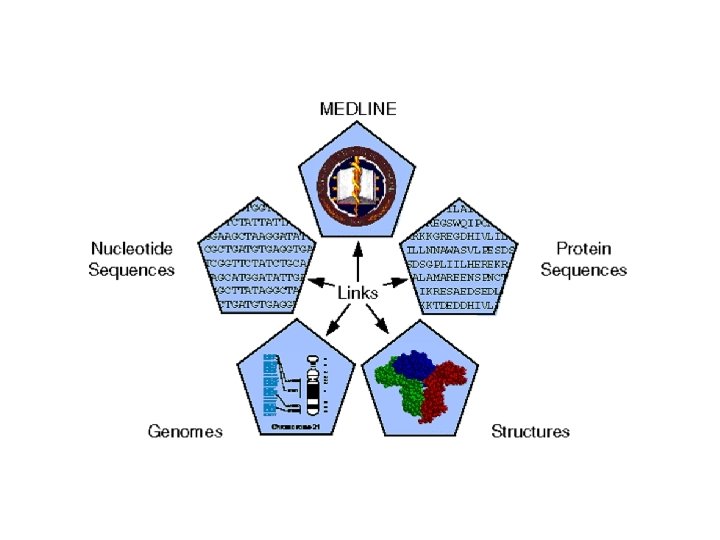

• These relationships might include genes in a multi-gene family, related journal articles, or other proteins in the same biochemical pathway • This potential for horizontal movement through the linked databases makes Entrez a dynamic tool. • You can start with only a vague set of keywords or a sequence from the laboratory and rapidly access a set of relevant literature and related database sequences.

Similarity Searching • There a variety of computer programs that are used for making comparisons between DNA sequences. • The most popular is known as BLAST (Basic Local Alignment Search Tool) • BLAST is free at the NCBI website

BLAST Searches Gen. Bank The NCBI BLAST web server lets you compare your query sequence to various sections of Gen. Bank – nr = non-redundant (main sections) – month = new sequences from the past few weeks – ESTs – human, drososphila, yeast, or E. coli genomes – proteins (by automatic translation) • This is a VERY fast and powerful computer.

BLAST is Complex • Similarity searching relies on the concepts of alignment and distance between pairs of sequences. • Distances can only be measured between aligned sequences (match vs. mismatch at each position). • A similarity search is a process of testing the best alignment of a query sequence with every sequence in a database.
 4 DNA bases vs. 20 amino acids -")
Search with Protein not DNA 1) 4 DNA bases vs. 20 amino acids - less random similarity 2) Can have varying degrees of similarity between different AAs - # of mutations, chemical similarity, PAM matrix 3) Protein databanks are much smaller than DNA databanks.
")
BLAST has Automatic Translation • BLASTX makes automatic translation (in all 6 reading frames) of your DNA query sequence to compare with protein databanks • TBLASTN makes automatic translation of an entire DNA database to compare with your protein query sequence • Only make a DNA-DNA search if you are working with a sequence that does not code for protein.

• • >gb|BE 588357. 1|BE 588357 194087 BARC 5 BOV Bos taurus c. DNA 5'. Length = 369 • Score = 272 bits (137), • • Identities = 258/297 (86%), Gaps = 1/297 (0%) Strand = Plus / Plus Expect = 4 e-71 • • • • • Query: 17 aggatccaacgtcgctccagctgctcttgacgactccacagataccccgaagccatggca 76 |||||||| || | ||||| Sbjct: 1 aggatccaacgtcgctgcggctacccttaaccact-cgcagaccccccgcagccatggcc 59 Query: 77 agcaagggcttgcaggacctgaagcaacaggtggaggggaccgcccaggaagccgtgtca 136 |||||||||||| | |||||| || Sbjct: 60 agcaagggcttgcaggacctgaagaagcaagtggagggggcggcccaggaagcggtgaca 119 Query: 137 gcggccggagcggcagctcagcaagtggtggaccaggccacagaggcggggcagaaagcc 196 |||| | ||||||||| || |||||| Sbjct: 120 tcggccggaacagcggttcagcaagtggtggatcaggccacagaagcagggcagaaagcc 179 Query: 197 atggaccagctggccaagaccacccaggaaaccatcgacaagactgctaaccaggcctct 256 ||||| | ||||||||||||| Sbjct: 180 atggaccaggttgccaagactacccaggaaaccatcgaccagactgctaaccaggcctct 239 Query: 257 gacaccttctctgggattgggaaaaaattcggcctcctgaaatgacagcagggagac 313 || || ||||||||| |||| Sbjct: 240 gagactttctcgggttttgggaaaaaacttggcctcctgaaatgacagaagggagac 296

Understand the Statistics! • BLAST produces an E-value for every match – This is the same as the P value in a statistical test • A match is generally considered significant if the E-value < 0. 05 (smaller numbers are more significant) • Very low E-values (e-100) are homologs or identical genes • Moderate E-values are related genes • Long regions of moderate similarity are more important than short regions of high identity.

BLAST is Approximate • BLAST makes similarity searches very quickly because it takes shortcuts. – looks for short, nearly identical “words” (11 bases) • It also makes errors – misses some important similarities – makes many incorrect matches • easily fooled by repeats or skewed composition

Bad Genome Annotation • Gene finding is at best only 90% accurate. • New sequences are automatically annotated with BLAST scores. • Bad annotations propagate • Its going to take us 10 -20 years or more to sort this mess out!

Protein Function • The ultimate goal of the HGP is to identify all of the genes and determine their functions • Genes function by being translated into proteins: – structural – enzymes – regulatory – signalling

Translation • Once we have found the DNA sequence of a gene, we can decode the amino acid sequence of the corresponding protein. • The “Genetic Code” is actually quite simple.

Chemical Properties Some chemical properties of a protein can be calculated from its amino acid sequence: • molecular weight • charge/p. H • hydrophobicity

Patterns in Proteins

Conserved Domains • Proteins are built out of functional units know as domains (or motifs) • These domains have conserved sequences Often much more similar than their respective proteins u Exon splicing theory (W. Gilbert) • Exons correspond to folding domains which in turn serve as functional units • Unrelated proteins may share a single similar exon (i. e. . ATPase or DNA binding function) u

Simple Structures Some motifs form structures that can be recognized as simple sequence patterns: – transmembrane domains – coiled coils – helix-turn-helix – signal peptides

Functional Motifs • Other functional portions of proteins can be recognized by their sequence, even if their 3 D structure is not known. • There are many databases of protein motifs/domains: Pro. Site, Pfam, Pro. Dom, etc.

Tools for Finding Motifs • Define a motif from a set of known proteins that share a similar sequence and function. • A pattern is a list of amino acids that can occur at each position in the motif. • A profile is a matrix that assigns a value to every amino acid at every position in the motif. • A HMM is a more complex profile based on pairs of amino acids.


Protein 3 -D Structure

Structure = Function • Proteins function by 3 -D interactions with other molecules (i. e. physical chemistry). • So for a protein, 3 -D structure is function. • But we can’t accurately determine 3 -D structure from gene sequence.

Structure Prediction Predicting a protein’s 3 -D structure from its amino acid sequence is incredibly complex. – proteins are polypeptides (long chains of amino acids) – can fold and rotate around bonds within each amino acid as well as the bonds between them – it is not possible to evaluate every possible folding pattern for an amino acid sequence

Secondary Structure • The local structure of the amino acids in a protein can also be predicted to some extent. • Each amino acid has a tendency to form either an alpha helix or a beta sheet

Threading • Rather than computing a 3 -D structure from scratch, it may be possible to find a similar structure. • Must have ~25% aa sequence identity. • Uses a process called threading to create a new structure based on a known structure. • This still requires HUGE amounts of computer power.
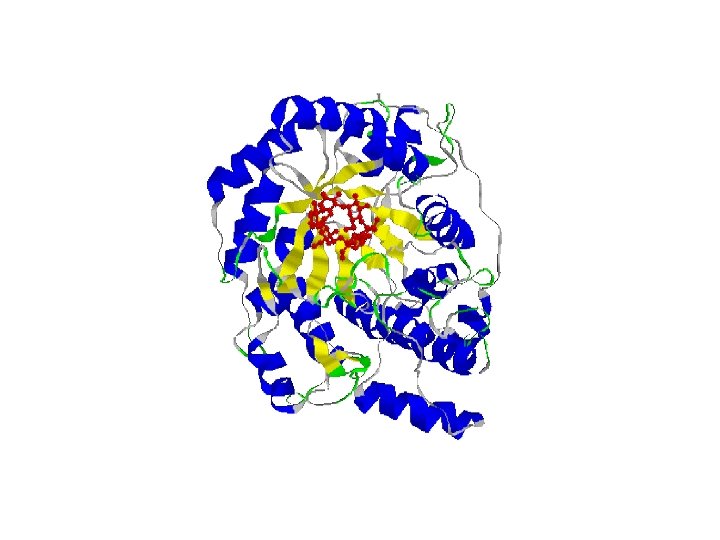

Protein Data Base • There is a database of all known protein structures called the PDB. • These have been determined by X-ray crystalography and/or NMR. • Anyone download and view these structures with a PDB viewer program.

Ras. Mol is the simplest PDB viewer. http: //www. umass. edu/microbio/rasmol/ It can work together with a web browser to let you view the structure of any sequence found with Entrez that has a known 3 -D structure.

Gene Finding & Translation • How can we find genes on chromosomes? • Genome project data is just huge chunks of DNA. • Does automatic annotation work?

Raw Genome Data:

Finding Genes is Not Easy • Perhaps 1% of human DNA encodes functional genes. • Genes are interspersed among long stretches of non-coding DNA. • Repeats, pseudo-genes, and introns confound matters

Pattern Finding Tools It is possible to use DNA sequence patterns to predict genes: • Promoters • translational start and stop codes (ORFs) • intron splice sites • codon usage
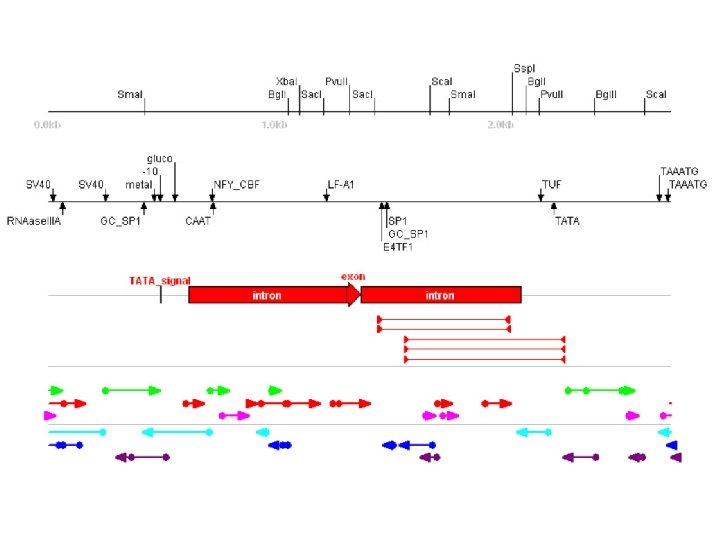

Similarity to Known Genes • It is also possible to scan new DNA sequence for known genes • Can look for annotated genes/proteins • Or just for RNAs (ESTs)
f2d95b5c2151e206cc4f5c773c232fa1.ppt