

апластические анемии.ppt
- Количество слайдов: 67
 Апластические анемии в детском возрасте д. м. н. Романова О. Н. , к. м. н. Мигаль Н. В. ГУ «РНПЦДОГ» , БГМУ, кафедра детских болезней № 1
Апластические анемии в детском возрасте д. м. н. Романова О. Н. , к. м. н. Мигаль Н. В. ГУ «РНПЦДОГ» , БГМУ, кафедра детских болезней № 1
 Апластическая анемия. ► АА – группа наследственных и приобретенных заболеваний, обусловленных дефектом стволовой клетки или ее микроокружения, приводящим к уменьшению или отсутствию продукции гемопоэтических клеток, жировым замещением костного мозга и, как следствие – к панцитопении в периферической крови
Апластическая анемия. ► АА – группа наследственных и приобретенных заболеваний, обусловленных дефектом стволовой клетки или ее микроокружения, приводящим к уменьшению или отсутствию продукции гемопоэтических клеток, жировым замещением костного мозга и, как следствие – к панцитопении в периферической крови
 Распространенность: Все АА - 1 -2 случая") Апластическая анемия. ► Приобретенная ► Врожденная (наследственная) Распространенность: Все АА - 1 -2 случая на 1 млн населения в год Приобретенные АА – 0, 2 -0, 6 на 100 000 детского населения в год Распределение по полу м: д = 1 : 1. В 2 - 3 раза чаще на Дальнем Востоке.
Апластическая анемия. ► Приобретенная ► Врожденная (наследственная) Распространенность: Все АА - 1 -2 случая на 1 млн населения в год Приобретенные АА – 0, 2 -0, 6 на 100 000 детского населения в год Распределение по полу м: д = 1 : 1. В 2 - 3 раза чаще на Дальнем Востоке.
 Классификация АА. Приобретенная АА 1. 2. ► ► ► Идиопатическая – 87% Вторичная: облучение; лекарства и химические вещества: (цитостатики, хлорамфеникол, противовоспалительные, противосудорж-ные, препараты золота, дезагреганты, бензин, промышленные яды и др. ); Вирусы: (вирусные гепатиты (флавовирус) – 6%, парвовирусы, ВЭБ, ветряная оспа и др. )
Классификация АА. Приобретенная АА 1. 2. ► ► ► Идиопатическая – 87% Вторичная: облучение; лекарства и химические вещества: (цитостатики, хлорамфеникол, противовоспалительные, противосудорж-ные, препараты золота, дезагреганты, бензин, промышленные яды и др. ); Вирусы: (вирусные гепатиты (флавовирус) – 6%, парвовирусы, ВЭБ, ветряная оспа и др. )
 Классификация АА. Наследственная АА ► анемия Фанкони; ► анемия Эстрена-Дамешека ► анемия Блекфана-Даймонда ► врожденный дискератоз; ► анемия Швахмана-Даймонда-Оски; ► ретикулярный дизгенез; ► амегакариоцитарная тромбоцитопения; ► семейные апластические анемии;
Классификация АА. Наследственная АА ► анемия Фанкони; ► анемия Эстрена-Дамешека ► анемия Блекфана-Даймонда ► врожденный дискератоз; ► анемия Швахмана-Даймонда-Оски; ► ретикулярный дизгенез; ► амегакариоцитарная тромбоцитопения; ► семейные апластические анемии;
 Классификация АА в зависимости от клеточной линии Угнетение красного ростка ► Приобретенная: идиопатическая; лекарства и токсины; иммуннная; тимома; транзиторная эритробластопения младенцев; ► Наследственная: анемия Даймонда Блекфана.
Классификация АА в зависимости от клеточной линии Угнетение красного ростка ► Приобретенная: идиопатическая; лекарства и токсины; иммуннная; тимома; транзиторная эритробластопения младенцев; ► Наследственная: анемия Даймонда Блекфана.
 Классификация АА в зависимости от клеточной линии Нейтропения ► Приобретенная: идиопатическая; лекарства и токсины; ► Наследственная: синдром Костмана; синдром Швахмана-Даймонда-Оски; ретикулярный дизгенез.
Классификация АА в зависимости от клеточной линии Нейтропения ► Приобретенная: идиопатическая; лекарства и токсины; ► Наследственная: синдром Костмана; синдром Швахмана-Даймонда-Оски; ретикулярный дизгенез.
 Классификация АА в зависимости от клеточной линии Тромбоцитопения ► Приобретенная: идиопатическая, лекарства и токсины. ► Наследственная: амегекариоцитарная, тромбоцитопения с отсутствием лучевой кости.
Классификация АА в зависимости от клеточной линии Тромбоцитопения ► Приобретенная: идиопатическая, лекарства и токсины. ► Наследственная: амегекариоцитарная, тромбоцитопения с отсутствием лучевой кости.
 Апластическая анемия. ► Первый случай АА был описан Эрлихом в 1888 году у беременной женщины. Заболевание характеризовалось тяжелой анемией, кровоизлиянием на коже и в склеру и высокой температурой. Исследование костного мозга при аутопсии выявило замещение кроветворных клеток жиром.
Апластическая анемия. ► Первый случай АА был описан Эрлихом в 1888 году у беременной женщины. Заболевание характеризовалось тяжелой анемией, кровоизлиянием на коже и в склеру и высокой температурой. Исследование костного мозга при аутопсии выявило замещение кроветворных клеток жиром.
 Апластическая анемия. ► Термин апластическая анемия был предложен в 1904 году Chauffard. ► Сниженная клеточность костного мозга может наблюдаться при АА, МДС, предлейкозах, лейкозах и лимфомах. ► Панцитопения может быть первичной и вторичной.
Апластическая анемия. ► Термин апластическая анемия был предложен в 1904 году Chauffard. ► Сниженная клеточность костного мозга может наблюдаться при АА, МДС, предлейкозах, лейкозах и лимфомах. ► Панцитопения может быть первичной и вторичной.
 Апластическая анемия. ► Первичная: МДС, ПНГ, миелофиброз, некоторые лейкозы и лимфомы, миелофтиз. ► Вторичная: системные заболевания (СКВ), гиперспленизм, дефицит В 12 и фолиевой кислоты, инфекции, алкоголизм, бруцеллез, саркоидоз, туберкулез. .
Апластическая анемия. ► Первичная: МДС, ПНГ, миелофиброз, некоторые лейкозы и лимфомы, миелофтиз. ► Вторичная: системные заболевания (СКВ), гиперспленизм, дефицит В 12 и фолиевой кислоты, инфекции, алкоголизм, бруцеллез, саркоидоз, туберкулез. .
 Апластическая анемия Лабораторная диагностика ► Общий анализ крови. Характерны снижение количества эритроцитов, гранулоцитов, тромбоцитов, ретикулоцитов. Анемия чаще нормоцитарная приобретенной АА, макроцитарная – при врожденных АА. Анализ крови необходимо делать в динамике, особенно в период становления заболевания. ► Миелограмма. Необходима пункция костного мозга минимум из трех анатомических точек, т. к. можно попасть в «горячий» карман (место, где сохранено кроветворение в полном объеме, особенно это касается грудины.
Апластическая анемия Лабораторная диагностика ► Общий анализ крови. Характерны снижение количества эритроцитов, гранулоцитов, тромбоцитов, ретикулоцитов. Анемия чаще нормоцитарная приобретенной АА, макроцитарная – при врожденных АА. Анализ крови необходимо делать в динамике, особенно в период становления заболевания. ► Миелограмма. Необходима пункция костного мозга минимум из трех анатомических точек, т. к. можно попасть в «горячий» карман (место, где сохранено кроветворение в полном объеме, особенно это касается грудины.
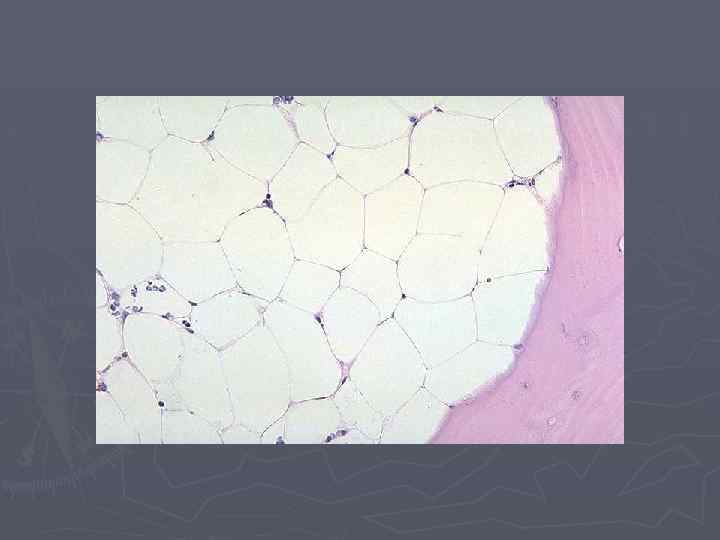
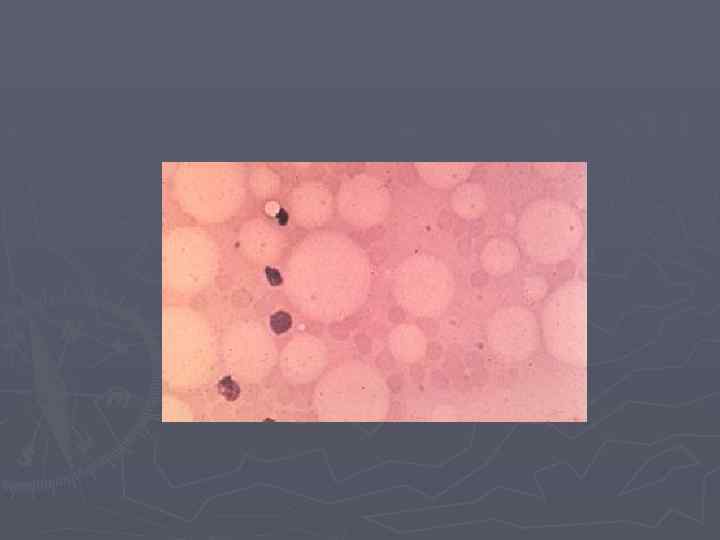
 Апластическая анемия Лабораторная диагностика ► Трепанобиопсия – определяет клеточность костного мозга (для достоверной оценки необходимо наличие 6 -9 костномозгоаых пространств) и степень замещения жировой и фиброзной тканью (> 75%). ► Цитогенетическое исследование костного мозга и периферической крови (проба с диэпокибутаном, выявляющую повышенную ломкость хромосом – подтверждает диагноз анемии Фанкони). ► Иммунологическое исследование клеток костного мозга (иммунофенотипирование) с определением СД 34+ клеток.
Апластическая анемия Лабораторная диагностика ► Трепанобиопсия – определяет клеточность костного мозга (для достоверной оценки необходимо наличие 6 -9 костномозгоаых пространств) и степень замещения жировой и фиброзной тканью (> 75%). ► Цитогенетическое исследование костного мозга и периферической крови (проба с диэпокибутаном, выявляющую повышенную ломкость хромосом – подтверждает диагноз анемии Фанкони). ► Иммунологическое исследование клеток костного мозга (иммунофенотипирование) с определением СД 34+ клеток.
 Апластическая анемия Лабораторная диагностика ► Титр РНК, ДНК, проба Кумбса, ревматоидный фактор, пробы печени, туберкулиновый тест. ► Детальный анамнез заболевания: токсины, облучение, лекарства, наследственные АА. ► Серологические исследования на вирусы в частности на вирус ГА, ГВ, ГС, ВИЧ, ВЭБ, парвовирус В 19 и др. ► Содержание В 12 в сыворотке и эритроцитах, фолиевой кислоты; ► Проба Хема. ► Скелетограмма и рентгенограмма.
Апластическая анемия Лабораторная диагностика ► Титр РНК, ДНК, проба Кумбса, ревматоидный фактор, пробы печени, туберкулиновый тест. ► Детальный анамнез заболевания: токсины, облучение, лекарства, наследственные АА. ► Серологические исследования на вирусы в частности на вирус ГА, ГВ, ГС, ВИЧ, ВЭБ, парвовирус В 19 и др. ► Содержание В 12 в сыворотке и эритроцитах, фолиевой кислоты; ► Проба Хема. ► Скелетограмма и рентгенограмма.
 Тяжелая форма Сверхтяжелая форма Гранулоциты<500/мкл Определяется только Тромбоциты<20000/мкл") Апластическая анемия. Критерии тяжести АА (Camitta) Тяжелая форма Сверхтяжелая форма Гранулоциты<500/мкл Определяется только Тромбоциты<20000/мкл числом гранулоцитов Ретикулоциты<40000/м <200/мкл кл
Апластическая анемия. Критерии тяжести АА (Camitta) Тяжелая форма Сверхтяжелая форма Гранулоциты<500/мкл Определяется только Тромбоциты<20000/мкл числом гранулоцитов Ретикулоциты<40000/м <200/мкл кл
 Приобретенная АА. ► Клиника: связана и зависит от тяжести панцитопении в ОАК. ► Геморрагический синдром обычно возникает первым и зависит от количества тромбоцитов. Проявляется петехиальной сыпью, экхимозами, носовыми кровотечениями, кровоточивостью слизистых. ► Нейтропения - причина язвенного стоматита, бактериальных инфекций, лихорадки. ► Анемия - причина бледности, утомляемости,
Приобретенная АА. ► Клиника: связана и зависит от тяжести панцитопении в ОАК. ► Геморрагический синдром обычно возникает первым и зависит от количества тромбоцитов. Проявляется петехиальной сыпью, экхимозами, носовыми кровотечениями, кровоточивостью слизистых. ► Нейтропения - причина язвенного стоматита, бактериальных инфекций, лихорадки. ► Анемия - причина бледности, утомляемости,
 Экспрессия аберрантного протеина гемопоэтической клеткой Захват") Патогенез приобретенной АА Антигенный стимул (вирус гепатита, медикамент) Экспрессия аберрантного протеина гемопоэтической клеткой Захват аберрантных протеинов антигенпрезентирующими клетками (макрофаги) с формированием пептидов
Патогенез приобретенной АА Антигенный стимул (вирус гепатита, медикамент) Экспрессия аберрантного протеина гемопоэтической клеткой Захват аберрантных протеинов антигенпрезентирующими клетками (макрофаги) с формированием пептидов
 Патогенез приобретенной АА Пептиды образуют комплекс с MHLA Комплекс взаимодействует с Т-лимфоцитами Иммунный ответ Гибель аберрантной антигенпрезентирующей гемопоэтической клетки
Патогенез приобретенной АА Пептиды образуют комплекс с MHLA Комплекс взаимодействует с Т-лимфоцитами Иммунный ответ Гибель аберрантной антигенпрезентирующей гемопоэтической клетки
 АПЛАСТИЧЕСКИЕ АНЕМИИ патофизиология Цитотоксические Т-лимфоциты Размер пула стволовых клеток время
АПЛАСТИЧЕСКИЕ АНЕМИИ патофизиология Цитотоксические Т-лимфоциты Размер пула стволовых клеток время
 Патогенез приобретенной АА
Патогенез приобретенной АА
 Приобретенная АА. ► Диагноз ПАА выставляется только при наличии следующих критериев: ► клеточность костного мозга < 25% от нормы (по данным трепанобиопсии); ► в ОАК: тромбоциты менее 20 000 /мм 3, гранулоциты менее 0, 5 х 10 9/л и ретикулоциты менее 20 х 10 9/л.
Приобретенная АА. ► Диагноз ПАА выставляется только при наличии следующих критериев: ► клеточность костного мозга < 25% от нормы (по данным трепанобиопсии); ► в ОАК: тромбоциты менее 20 000 /мм 3, гранулоциты менее 0, 5 х 10 9/л и ретикулоциты менее 20 х 10 9/л.
 Приобретенная АА. ► Дифференциальная диагностика: ► МДС; ► лейкозы; ► миелофиброз; ► врожденная анемия Фанкони; ► лимфомы, ► волосатоклеточный лейкоз.
Приобретенная АА. ► Дифференциальная диагностика: ► МДС; ► лейкозы; ► миелофиброз; ► врожденная анемия Фанкони; ► лимфомы, ► волосатоклеточный лейкоз.
 Приобретенная АА. ► Минимальное время наблюдения до начала терапии: при СТАА и ТАА - 2 недели; для НТАА - 6 недель. ► Лечение АА проводится только в специализированном гематологическом отделении
Приобретенная АА. ► Минимальное время наблюдения до начала терапии: при СТАА и ТАА - 2 недели; для НТАА - 6 недель. ► Лечение АА проводится только в специализированном гематологическом отделении
 Приобретенная АА. Лечение приобретенной АА ► наличие HLA - идентичного родственного донора – ТКМ. Поэтому до начала лечения желательно HLA-типирование больного и членов семьи. ► при отсутствии донора – иммуносупрессивная терапия (ИСТ).
Приобретенная АА. Лечение приобретенной АА ► наличие HLA - идентичного родственного донора – ТКМ. Поэтому до начала лечения желательно HLA-типирование больного и членов семьи. ► при отсутствии донора – иммуносупрессивная терапия (ИСТ).
 Приобретенная АА. Возможности лечения рефрактерной анемии ► Андрогены. Наиболее часто используют 3 -х месячный курс даназола или декадураболина. В результате их применения имеются данные о исчезновении потребности в трансфузиях эритроцитов. ► Применение факторов роста. ► Совместимая неродственная ТКМ. Риск возникновения РТПХ увеличивается. Долгосрочная выживаемость у детей составляет 50%, у пациентов более старшего возраста - еще меньше.
Приобретенная АА. Возможности лечения рефрактерной анемии ► Андрогены. Наиболее часто используют 3 -х месячный курс даназола или декадураболина. В результате их применения имеются данные о исчезновении потребности в трансфузиях эритроцитов. ► Применение факторов роста. ► Совместимая неродственная ТКМ. Риск возникновения РТПХ увеличивается. Долгосрочная выживаемость у детей составляет 50%, у пациентов более старшего возраста - еще меньше.
 Приобретенная парциальная красноклеточная аплазия. ► ПККА проявляется анемией, ретикулоцитопенией и отсутствием костномозговых предшественников. ► Эпидемиология: редко встречается, ж : м = 2 : 1. ► Средний возраст начала заболевания около 60 лет.
Приобретенная парциальная красноклеточная аплазия. ► ПККА проявляется анемией, ретикулоцитопенией и отсутствием костномозговых предшественников. ► Эпидемиология: редко встречается, ж : м = 2 : 1. ► Средний возраст начала заболевания около 60 лет.
 Приобретенная парциальная красноклеточная аплазия. ► Этиология. Тимома, злокачественные опухоли лимфоидной ткани, ХМЛ, МДС, миелофиброз, сосудистые коллагенозы, беременность, паранеопластические синдромы, вирусы и лекарственные препараты.
Приобретенная парциальная красноклеточная аплазия. ► Этиология. Тимома, злокачественные опухоли лимфоидной ткани, ХМЛ, МДС, миелофиброз, сосудистые коллагенозы, беременность, паранеопластические синдромы, вирусы и лекарственные препараты.
 Приобретенная парциальная красноклеточная аплазия. ► Диагностика: отличительными признаками ПККА является анемия, ретикулоцитопения и изолированный дефицит эритробластов в костном мозге. ► Диагностика включает: ОАК + формула + ретикулоциты + морфология эритроцитов; исследование костного мозга (изолированное поражение красного ростка с нормальной клеточностью. Мегакариоцитарный и гранулоцитарный ростки сохранены). ► Исследование на парвовирусы (серология), КТГ грудной клетки.
Приобретенная парциальная красноклеточная аплазия. ► Диагностика: отличительными признаками ПККА является анемия, ретикулоцитопения и изолированный дефицит эритробластов в костном мозге. ► Диагностика включает: ОАК + формула + ретикулоциты + морфология эритроцитов; исследование костного мозга (изолированное поражение красного ростка с нормальной клеточностью. Мегакариоцитарный и гранулоцитарный ростки сохранены). ► Исследование на парвовирусы (серология), КТГ грудной клетки.
 Приобретенная парциальная красноклеточная аплазия. Дифференциальная диагностика: 1. Наследственная ПККА: анемия Даймонда. Блекфана. 2. Неиммунная гидроцефалия плода: внутриутробное инфицирование парвовирусом В 12. 3. Транзиторные синдромы: транзиторная детская эритробластпения, транзиторный апластический гемолитический криз. ►
Приобретенная парциальная красноклеточная аплазия. Дифференциальная диагностика: 1. Наследственная ПККА: анемия Даймонда. Блекфана. 2. Неиммунная гидроцефалия плода: внутриутробное инфицирование парвовирусом В 12. 3. Транзиторные синдромы: транзиторная детская эритробластпения, транзиторный апластический гемолитический криз. ►
 Приобретенная парциальная красноклеточная аплазия. ► 1. 2. 3. Лечение. Необходимо прекратить прием лекарств, воздействие которых увеличивает риск развития цитопении. . Парвовирус В 12. Эффективно назначение иммуноглобулинов внутривенно. Тимома. Проводится хирургическое лечение.
Приобретенная парциальная красноклеточная аплазия. ► 1. 2. 3. Лечение. Необходимо прекратить прием лекарств, воздействие которых увеличивает риск развития цитопении. . Парвовирус В 12. Эффективно назначение иммуноглобулинов внутривенно. Тимома. Проводится хирургическое лечение.
 Приобретенная парциальная красноклеточная аплазия. ► 1. 2. Лечение. Аутоиммунная ПККА. Назначается поэтапная иммуносуперссивная терапия до достижения ремиссии или доисчерпывания терапевтических возможностей. Начинают с наиболее щадящих методов. Преднизолон. Азатиоприн или циклофосфан (внутрь) +/- преднизолон, постепенно увеличивать дозу азатиоприна циклофосфана до тех пор пока: а) количество ретикулоцитов не возрастает; б) количество лейкоцитов не снизится менее 2000/мкл; в) количество тромбоцитов не снизится менее 80. 000/мкл.
Приобретенная парциальная красноклеточная аплазия. ► 1. 2. Лечение. Аутоиммунная ПККА. Назначается поэтапная иммуносуперссивная терапия до достижения ремиссии или доисчерпывания терапевтических возможностей. Начинают с наиболее щадящих методов. Преднизолон. Азатиоприн или циклофосфан (внутрь) +/- преднизолон, постепенно увеличивать дозу азатиоприна циклофосфана до тех пор пока: а) количество ретикулоцитов не возрастает; б) количество лейкоцитов не снизится менее 2000/мкл; в) количество тромбоцитов не снизится менее 80. 000/мкл.
 Анемия Фанкони. Характеризуется: ► аутосомно-рецессивным типом наследования; ► наличием врожденных аномалий ► прогрессирующей аплазией костного мозга ► предрасположенностью к злокачественным новообразованиям (чаще МДС и ОМЛ, в белее зрелом возрасте - чешуйчатые карциномы головы, шеи и женской половой сферы)
Анемия Фанкони. Характеризуется: ► аутосомно-рецессивным типом наследования; ► наличием врожденных аномалий ► прогрессирующей аплазией костного мозга ► предрасположенностью к злокачественным новообразованиям (чаще МДС и ОМЛ, в белее зрелом возрасте - чешуйчатые карциномы головы, шеи и женской половой сферы)
 Анемия Фанкони врожденное аутосомно-рецессивное заболевание, характеризующееся нестабильностью геномного аппарата Впервые описано Гвидо Фанкони в 1927 году
Анемия Фанкони врожденное аутосомно-рецессивное заболевание, характеризующееся нестабильностью геномного аппарата Впервые описано Гвидо Фанкони в 1927 году
 Анемия Фанкони демография Заболеваемость ~ 1 случай на 1 миллион населения Носительство генов ~ 1 на 300 человек • средний возраст презентации - 8 лет (0 - 48) • средняя продолжительность жизни - 16 лет • с момента развития аплазии ~ 5 лет • причины смерти: костно-мозговая недостаточность, опухоли
Анемия Фанкони демография Заболеваемость ~ 1 случай на 1 миллион населения Носительство генов ~ 1 на 300 человек • средний возраст презентации - 8 лет (0 - 48) • средняя продолжительность жизни - 16 лет • с момента развития аплазии ~ 5 лет • причины смерти: костно-мозговая недостаточность, опухоли
 Анемия Фанкони клинические проявления костно-мозговая недостаточность ► множественные соматические аномалии ► высокий риск развития опухолей ►
Анемия Фанкони клинические проявления костно-мозговая недостаточность ► множественные соматические аномалии ► высокий риск развития опухолей ►
 Врожденные АА Анемия Фанкони
Врожденные АА Анемия Фанкони
 АНЕМИЯ ФАНКОНИ
АНЕМИЯ ФАНКОНИ
 Анемия Фанкони клинические проявления Соматические аномалии и пороки развития ► аномальная пигментация кожи ► задержка роста ► пороки костей конечностей, лицевого скелета ► аномалии глаз, ушей ► аномалии внутренних органов: мочеполовых, сердечно-легочные, желудочно-кишечные ► без аномалий 10 - 15% больных АФ
Анемия Фанкони клинические проявления Соматические аномалии и пороки развития ► аномальная пигментация кожи ► задержка роста ► пороки костей конечностей, лицевого скелета ► аномалии глаз, ушей ► аномалии внутренних органов: мочеполовых, сердечно-легочные, желудочно-кишечные ► без аномалий 10 - 15% больных АФ
 Анемия Фанкони
Анемия Фанкони
 Анемия Фанкони Клинические проявления Костно-мозговая недостаточность темпы начала и прогрессирования варьируют гипорегенераторная макроцитарная нормохромная анемия тромбоцитопения лейкопения
Анемия Фанкони Клинические проявления Костно-мозговая недостаточность темпы начала и прогрессирования варьируют гипорегенераторная макроцитарная нормохромная анемия тромбоцитопения лейкопения
 Анемия Фанкони клинические проявления Опухоли злокачественные новообразования - острый миелолейкоз / МДС, солидные опухоли к 20 годам у 20% больных АФ к 45 годам у 75% больных АФ солидные опухоли в возрасте 15 лет Риск опухолей у членов семьи б-х АФ составляет 25% B. Alter et al. , M. Ferrara et al, Blood 2001, v. 98, № 11
Анемия Фанкони клинические проявления Опухоли злокачественные новообразования - острый миелолейкоз / МДС, солидные опухоли к 20 годам у 20% больных АФ к 45 годам у 75% больных АФ солидные опухоли в возрасте 15 лет Риск опухолей у членов семьи б-х АФ составляет 25% B. Alter et al. , M. Ferrara et al, Blood 2001, v. 98, № 11
 Субтипы Анемии Фанкони Субтип A B C D E %АФ 66 4. 3 12. 7 Хромосома 16 q 24. 3 ? 9 q 22. 3 3 p 22 -26 ? Белок 163 k. Da -
Субтипы Анемии Фанкони Субтип A B C D E %АФ 66 4. 3 12. 7 Хромосома 16 q 24. 3 ? 9 q 22. 3 3 p 22 -26 ? Белок 163 k. Da -
 Анемия Фанкони. ► Для подтверждения АФ необходимо провести тест на ломкость хромосом с диэпоксибутаном или митомицином С. ► Гематологические проявления АФ могут варьировать от нормального количества клеток крови с небольшим уровнем гемоглобина F до тяжелой панцитопении с полной зависимостью больного от
Анемия Фанкони. ► Для подтверждения АФ необходимо провести тест на ломкость хромосом с диэпоксибутаном или митомицином С. ► Гематологические проявления АФ могут варьировать от нормального количества клеток крови с небольшим уровнем гемоглобина F до тяжелой панцитопении с полной зависимостью больного от
 АНЕМИЯ ФАНКОНИ Цитогенетические тесты Характерные хромосомные аберрации у больных АФ: разрывы хромосом, перестройки, эндоредупликации, обмены, сшивки, кольцевые хромосомы.
АНЕМИЯ ФАНКОНИ Цитогенетические тесты Характерные хромосомные аберрации у больных АФ: разрывы хромосом, перестройки, эндоредупликации, обмены, сшивки, кольцевые хромосомы.
 АНЕМИЯ ФАНКОНИ Цитогенетические тесты
АНЕМИЯ ФАНКОНИ Цитогенетические тесты
 АНЕМИЯ ФАНКОНИ Цитогенетические тесты
АНЕМИЯ ФАНКОНИ Цитогенетические тесты
 Анемия Фанкони Этиология и патогенез Мутации гена FANC A
Анемия Фанкони Этиология и патогенез Мутации гена FANC A
 Патогенез Анемии Фанкони ► Длительность G 2 фазы клеточного цикла ► чувствительность к кислороду ► чувствительность к радиации ► Гиперпродукция TNFα ► Дефект способности восстановления ДНК ► Нестабильность генома повышенная ломкость хромосом ► Повышенный апоптоз ► Дефектная индукция Р 53
Патогенез Анемии Фанкони ► Длительность G 2 фазы клеточного цикла ► чувствительность к кислороду ► чувствительность к радиации ► Гиперпродукция TNFα ► Дефект способности восстановления ДНК ► Нестабильность генома повышенная ломкость хромосом ► Повышенный апоптоз ► Дефектная индукция Р 53
 Врожденные АА Анемия Фанкони Диагноз На основании гиперчувствительности лимфоцитов к кластогенным агентам Диэпоксибутан препараты платины Митомицин С
Врожденные АА Анемия Фанкони Диагноз На основании гиперчувствительности лимфоцитов к кластогенным агентам Диэпоксибутан препараты платины Митомицин С
 Врожденные АА Анемия Фанкони Лечение Консервативное Андрогены Ответ ~ 50% Медиана выживаемости 9 лет ТКМ Тестировать донора! Низкие дозы Cph Fludara Выживаемость 70% Опухоли!
Врожденные АА Анемия Фанкони Лечение Консервативное Андрогены Ответ ~ 50% Медиана выживаемости 9 лет ТКМ Тестировать донора! Низкие дозы Cph Fludara Выживаемость 70% Опухоли!
 Анемия Фанкони. ► гемотрансфузий. ► Лечение: андрогены, которые эффективны у 50% пациентов. Вначале реагирует эритроцитарный росток, затем гранулоцитарный, последним повышается количество тромбоцитов. ► Радикальным методом лечения является ТКМ, которая приводит к выздоровлению у 2/3 больных.
Анемия Фанкони. ► гемотрансфузий. ► Лечение: андрогены, которые эффективны у 50% пациентов. Вначале реагирует эритроцитарный росток, затем гранулоцитарный, последним повышается количество тромбоцитов. ► Радикальным методом лечения является ТКМ, которая приводит к выздоровлению у 2/3 больных.
 Анемия Даймонда - Блекфана. ► Диагностическими критериями являются: ретикулоцитопения с макроцитарной или нормоцитарной анемией; нормальная клеточность костного мозга с изолированной эритройдной гипоплазией; нормальное или близко к нормальному количество лейкоцитов и тромбоцитов.
Анемия Даймонда - Блекфана. ► Диагностическими критериями являются: ретикулоцитопения с макроцитарной или нормоцитарной анемией; нормальная клеточность костного мозга с изолированной эритройдной гипоплазией; нормальное или близко к нормальному количество лейкоцитов и тромбоцитов.
 Анемия Даймонда Блекфана. ► Развивается спорадически, хотя в различных семьях зарегистрированы аутосомно-рецессивный или аутосомно- доминантный тип наследования. ► Как правило первые признаки появляются в первые 6 месяцев жизни, в крайнем случае - после 1 года. ► Физические аномалии выявляются почти всегда у 25% детей.
Анемия Даймонда Блекфана. ► Развивается спорадически, хотя в различных семьях зарегистрированы аутосомно-рецессивный или аутосомно- доминантный тип наследования. ► Как правило первые признаки появляются в первые 6 месяцев жизни, в крайнем случае - после 1 года. ► Физические аномалии выявляются почти всегда у 25% детей.
 Генетика Анемии Даймонда. Блэкфана 1. Доминантное наследование ► Частая встречаемость заболевания у близких родственников ► Лабораторные тесты для исключения доминантного типа наследования: Hb, MCV, активность аденозин диаминазы в эритроцитах 2. Рецессивное наследование 3. Х-сцепленное наследование
Генетика Анемии Даймонда. Блэкфана 1. Доминантное наследование ► Частая встречаемость заболевания у близких родственников ► Лабораторные тесты для исключения доминантного типа наследования: Hb, MCV, активность аденозин диаминазы в эритроцитах 2. Рецессивное наследование 3. Х-сцепленное наследование
 Патогенез Анемии Даймонда. Блэкфана Патология клеток-предшественников эритропоэза, характеризующаяся: ► сниженной чувствительностью к EPO ► сниженная чувствительность к EPO не коррегируется IL-3 и GM-CSF ► 25% пациентов АДБ имеют мутацию гена, картированного на 19 q 23, кодирующего рибосомальный протеин S 19 (RPS 19)
Патогенез Анемии Даймонда. Блэкфана Патология клеток-предшественников эритропоэза, характеризующаяся: ► сниженной чувствительностью к EPO ► сниженная чувствительность к EPO не коррегируется IL-3 и GM-CSF ► 25% пациентов АДБ имеют мутацию гена, картированного на 19 q 23, кодирующего рибосомальный протеин S 19 (RPS 19)
 Клиника. Анемия Даймонда Блекфана ► Начинается у детей 1 -го года жизни ( нередко у новорожденных ► Ведущий клинический синдром - тяжелая гипорегенераторная анемия ► Задержка физического развития при нормальном психомоторном статусе ► Могут быть аномалии скелета, реже другие пороки развития ► Характерны «кофейные пятна» на коже, связанные с отложением меланина
Клиника. Анемия Даймонда Блекфана ► Начинается у детей 1 -го года жизни ( нередко у новорожденных ► Ведущий клинический синдром - тяжелая гипорегенераторная анемия ► Задержка физического развития при нормальном психомоторном статусе ► Могут быть аномалии скелета, реже другие пороки развития ► Характерны «кофейные пятна» на коже, связанные с отложением меланина
 Анемия Даймонда Блекфана. ► Лечение. ► Глюкокортикоиды (1 -3 мг/кг в сутки, критерии подбора дозы - независимость от гемотрансфузий), длительность приема регулируется индивидуально. ► Пульс-терапия - метипредом в/венно (максимальная доза - 30 мг/кг на 3 дня, далее 20 мг/кг - 3 дня, 10 мг/кг - 3 дня, затем переход на таблетированную форму в дозе 5 мг/кг с постепенным снижением дозы до поддерживающей). Эффект оценивают через 2 -3 мес.
Анемия Даймонда Блекфана. ► Лечение. ► Глюкокортикоиды (1 -3 мг/кг в сутки, критерии подбора дозы - независимость от гемотрансфузий), длительность приема регулируется индивидуально. ► Пульс-терапия - метипредом в/венно (максимальная доза - 30 мг/кг на 3 дня, далее 20 мг/кг - 3 дня, 10 мг/кг - 3 дня, затем переход на таблетированную форму в дозе 5 мг/кг с постепенным снижением дозы до поддерживающей). Эффект оценивают через 2 -3 мес.
 Анемия Даймонда Блекфана. Лечение: ► Рефрактерным пациентам показана долгосрочная трансфузия эритроцитов под контролем обмена железа. При показателях ферритина выше 1500 нг/мл показана терапия десфералом ► При стойкой ретикулоцитопении глюкокортикоидная терапия сочетается с препаратами фолиевой кислоты и витамина В 12
Анемия Даймонда Блекфана. Лечение: ► Рефрактерным пациентам показана долгосрочная трансфузия эритроцитов под контролем обмена железа. При показателях ферритина выше 1500 нг/мл показана терапия десфералом ► При стойкой ретикулоцитопении глюкокортикоидная терапия сочетается с препаратами фолиевой кислоты и витамина В 12
. Проявления синдрома включает изолированную анемию,") Транзиторная детская эритрбластпения. ► Причиной часто являются вирусы (парвовирусы). Проявления синдрома включает изолированную анемию, ретикулоцитопению и при исслдеовании костного мозга глубокую гипоплазию эритроидного ростка. Первые признаки ТДЭ обычно наблюдаются у детей старше 2 лет. В течение острой фазы
Транзиторная детская эритрбластпения. ► Причиной часто являются вирусы (парвовирусы). Проявления синдрома включает изолированную анемию, ретикулоцитопению и при исслдеовании костного мозга глубокую гипоплазию эритроидного ростка. Первые признаки ТДЭ обычно наблюдаются у детей старше 2 лет. В течение острой фазы
 Транзиторная детская эритрбластпения. ► ТДЭ, ретикулоцитопенической, маркеры напряжения эритропоэза отсутствуют, к которым относятся - увеличение среднего объема эритроцита, уровня фетального гемоглобина и уровня антигена I. Диагноз устанавливается часто ретроспективно. Нормализация гемопоза происходит в течение 1 - 2 мес.
Транзиторная детская эритрбластпения. ► ТДЭ, ретикулоцитопенической, маркеры напряжения эритропоэза отсутствуют, к которым относятся - увеличение среднего объема эритроцита, уровня фетального гемоглобина и уровня антигена I. Диагноз устанавливается часто ретроспективно. Нормализация гемопоза происходит в течение 1 - 2 мес.
 Дифференциальный диагноз ТДЭ и АДБ Признак Встречаемость Возраст ТДЭ АДБ часто 6 мес. - 4 года приобретенная редко 90% < 6 мес. генетическая нет есть (10 - 20%) вирус нет 25 % Длительность 1 нед. - 1 мес. пожизненно Трансфузии не зависимы МСV < 85μm 3 > 90μm 3 Hb. F норма повышен нет повышена ТКМ, гормоны, посиндромное Этиология Наследственность Анамнез Врожденные особенности AD эритроцитов Лечение посиндромное
Дифференциальный диагноз ТДЭ и АДБ Признак Встречаемость Возраст ТДЭ АДБ часто 6 мес. - 4 года приобретенная редко 90% < 6 мес. генетическая нет есть (10 - 20%) вирус нет 25 % Длительность 1 нед. - 1 мес. пожизненно Трансфузии не зависимы МСV < 85μm 3 > 90μm 3 Hb. F норма повышен нет повышена ТКМ, гормоны, посиндромное Этиология Наследственность Анамнез Врожденные особенности AD эритроцитов Лечение посиндромное
 Врожденный дискератоз Редкое заболевание, наследуется как: ► Х-сцепленный вариант в 75% случаев ► аутосомно-рецессивный вариант в 13% случаев ► аутосомно-доминантный вариант в 12% случаев ► Мальчики болеют чаще в 14 раз, чем девочки ► АА развивается у 50% больных. Дебют в среднем наступает в 19 лет (0, 5 -47 лет) ► Иногда начинается как изолированная тромбоцитопения (1 -5 лет), которая в дальнейшем трансформируется в панцитопению ► Типичны ретикулярная гиперпигментация, слезотечение, дистрофия ногтей (чаще на ногах), лейкоплакия слизистых рта
Врожденный дискератоз Редкое заболевание, наследуется как: ► Х-сцепленный вариант в 75% случаев ► аутосомно-рецессивный вариант в 13% случаев ► аутосомно-доминантный вариант в 12% случаев ► Мальчики болеют чаще в 14 раз, чем девочки ► АА развивается у 50% больных. Дебют в среднем наступает в 19 лет (0, 5 -47 лет) ► Иногда начинается как изолированная тромбоцитопения (1 -5 лет), которая в дальнейшем трансформируется в панцитопению ► Типичны ретикулярная гиперпигментация, слезотечение, дистрофия ногтей (чаще на ногах), лейкоплакия слизистых рта
 Врожденный дискератоз Лечение малоэффективно. ► Андрогены - эффективны в исключительных случаях ► ТКМ - остается очень проблематичной, т. к. часто впоследствии развивается цирроз печени, веноокклюзивная болезнь, поражение соединительной ткани ► Ростовые факторы - не эффективны
Врожденный дискератоз Лечение малоэффективно. ► Андрогены - эффективны в исключительных случаях ► ТКМ - остается очень проблематичной, т. к. часто впоследствии развивается цирроз печени, веноокклюзивная болезнь, поражение соединительной ткани ► Ростовые факторы - не эффективны
 Амегакариоцитарная тромбоцитопения ► Характерен дебют в виде изолированной тромбоцитопении в раннем возрасте ( до 1 года), далее быстро прогрессирует 3 -х ростковая цитопения. Часть больных имеют скелетные аномалии, эти дебютируют в более раннем возрасте. ► Лечение: очень эффективна ТКМ
Амегакариоцитарная тромбоцитопения ► Характерен дебют в виде изолированной тромбоцитопении в раннем возрасте ( до 1 года), далее быстро прогрессирует 3 -х ростковая цитопения. Часть больных имеют скелетные аномалии, эти дебютируют в более раннем возрасте. ► Лечение: очень эффективна ТКМ
 Синдром Швахмана-Даймонда. Оски ► Проявляется эндокринной недостаточность поджелудочной железы в сочетании с нейтропенией. У 25% развивается панцитопения. ► Мальчики болеют в 2 раза чаще, чем девочки ► Характерны появление еще в период новорожденности симптомов мальабсорбции, стеатореи (снижены или отсутствуют амилаза, липаза, трипсин). ► Нередко отмечается замедление развития, стигмы и аномалии развития (синдактилия, страбизм, короткая шея, гипертелоризм, незаращенное небо). ► Панцитопения, как правило, умеренная
Синдром Швахмана-Даймонда. Оски ► Проявляется эндокринной недостаточность поджелудочной железы в сочетании с нейтропенией. У 25% развивается панцитопения. ► Мальчики болеют в 2 раза чаще, чем девочки ► Характерны появление еще в период новорожденности симптомов мальабсорбции, стеатореи (снижены или отсутствуют амилаза, липаза, трипсин). ► Нередко отмечается замедление развития, стигмы и аномалии развития (синдактилия, страбизм, короткая шея, гипертелоризм, незаращенное небо). ► Панцитопения, как правило, умеренная