Апл анемии08.ppt
- Количество слайдов: 21


 - сборная группа заболеваний признаком которых является депрессия костномозгового")
АПЛАСТИЧЕСКИЕ АНЕМИИ Апластические анемии (АА) - сборная группа заболеваний признаком которых является депрессия костномозгового кроветворения (неспособность вырабатывать за единицу времени необходимое количество форменных элементов крови). При АА имеется утрата всех ростков гемопоэза: эритроидного, миелоидного, мегакариоцитарного; и замещение кроветворного костного мозга жировой тканью.

При поражении трех ростков кроветворения клинически заболевание характеризуется : Ø Ø Ø Анемией Геморрагическим синдромом Панцитопенией (анемия, тромбоцитопения, лейкоцитопения, ретикулоцитопения) Частыми инфекционными, септическими осложнениями Нет увеличения органов ретикулоэндотелиальной системы (печень селезёнка, лимфоузлы)

Заболеваемость Впервые заболевание было описано П. Эрлихом в 1888 году под названием панмиелофтиз. Частота у детей составляет 6 -10 случаев на 1000000 детского населения в год

ЭТИОЛОГИЯ АА является полиэтиологичным заболеванием Ø Ø Ø Экзогенные факторы I Физические 1. Ионизирующая радиация 2. Токи высокой частоты 3. Вибрация Ø Эндогенные факторы I Наследственные и генетические нарушения Ø II Нарушение функции желёз внутренний секреции: Ø Ø II Химические Ø * щитовидной железы Ø 1. Миелотоксические вещества: Ø Ø *Бензил, бензол и их производные *Пары ртути, азотной кислоты *Сернистый газ *Краски, лаки, нитроэмали *Пестициды, угольная и цинковая пыль *яичников *вилочковой железы Ø 2. Медикаменты Ø Ø *А/б (левомицетин, пенициллин, тетрациклин, макролиды) *Производные пиразолона (анальгин, амидопирин, бутадион) *Сульфаниламиды *Органические препараты мышьяка (новарсенол) *Антитиреоидные препараты (карбимазол) *НПВС (фенилбутазон) Ø Ø Ø Ø Ø III Системные соединительной ткани: Ø *системная красная волчанка *ревматоидный артрит *синдром Шегрена Ø Ø Ø Ø заболевания IV Беременность V Стрессы VI Травмы VII Пароксизмальная гемоглобинурия VIII Нарушения питания *квашиоркор *маразм ночная
 Ø Экзогенные факторы Ø Ø Ø *Органические препараты мышьяка (новарсенол) * Противотуберкулёзные")
ЭТИОЛОГИЯ (продолжение) Ø Экзогенные факторы Ø Ø Ø *Органические препараты мышьяка (новарсенол) * Противотуберкулёзные препараты ( стрептомицин. ПАСК, фтивазид) *Противогрибковые препараты (амфотерицин В) *Фенитоин противосудорожное, антиаритмик *Противомалярийные препараты (акрихин) * Противоопухолевые препараты (бусульфан) III Инфекционные Ø Ø 1. Вирусы Ø Ø *Гепатита А, В, С *Гриппа, парагриппа *Краснухи, кори *Эпидемического паротита Инфекционного мононуклеоза *Иммунодефицита человека *Цитомегаловирус (у новорожденных) *Вирус герпеса Ø 2. Бактерии Ø *Микобактерия туберкулеза Ø 3. Грибы Ø Ø Ø

По классификации В. И. Калиничевой выделяют следующие варианты болезни Ø I Наследственные апластические анемии Ø 1. Наследственные апластические анемии с общим поражением гемопоэза Ø 1. 1 Наследственная апластическая анемия с общим поражением гемопоэза и врожденными аномалиями развития (анемия Фанкони) Ø 1. 2 Наследственная семейная апластическая анемия с общим поражением гемопоэза без врожденных аномалий развития (анемия Эстрена-Дамешека) Ø 1. 3 Наследственная парциальная апластическая анемия с избирательным поражением эритропоэза (анемия Блекфена. Даймонда)

II Приобретенные апластические анемии Ø 1 С общим поражением гемопоэза Ø 1. 1 Острая апластическая анемия Ø 1. 2 Подострая апластическая анемия Ø 1. 3 Хроническая апластическая анемия Ø 2. С избирательным поражением эритропоэза (парциальная, чистая приобретенная красноклеточная апластическая анемия)

Согласно международным критериям оценки тяжести АА, выделяют 3 группы: Сверхтяжелая форма: в периферической крови глубокая панцитопения с быстрым и резким снижением гранулоцитов до единичных клеток в мазке крови и ретикулоцитов до полного исчезновения. Ø В костном мозге количество миелокариоцитов снижено менее 15*109/л, отсутствуют пролиферирующие элементы миелопоэза, содержание немиелоидных клеток более 80%. Значительно повышено количество плазматических клеток (до 8%) Ø В биоптате подвздошной кости определяется только жировая ткань. Ø

Ø Тяжелая форма: в периферической крови глубокое угнетение гемопоэза. Содержание гранулоцитов менее 0. 5*109/л, ретикулоцитов менее 1 количество миелокариоцитов менее 40*109/л, содержание немиелоидных клеток более 50%. ØВ пунктате костного мозга признаки относительно сохраненного гемопоэза в виде пролиферирующих элементов гранулоцитарного и эритроидного ростков.

Ø Нетяжелая форма: менее глубокое угнетение кроветворения. Количество гранулоцитов выше 0. 5*109/л, ретикулоцитов более 1%о. Ø Наряду с участками пониженной клеточности костного мозга встречаются нормоклеточные очаги, при этом количество немиелоидных клеток менее 50%, а эритроидных нормальное или повышенное.

ПЛАН ОБСЛЕДОВАНИЯ БОЛЬНЫХ АПЛАСТИЧЕСКИМИ АНЕМИЯМИ Ø Ø Ø Ø Клинический анализ крови, Гематокрит, Группа крови и резус фактор, Миелограммы из трех анатомически различных точек, определение колониеобразующих свойств и цитогенетический анализ при наследственных вариантах, Иммунологическое обследование: АТ к эритроцитам, тромбоцитам, лейкоцитам, Ig, типирование по HLA, РБТЛ. Биохимия крови: АЛТ, АСТ, билирубин, общий белок, протеинограмма, мочевина, креатинин, сахар, гаптоглобин, фетальный гемоглобин. Общеклиническое обследование: анализ мочи, копрограмма, посев кала, мазки из зева и носа, ЭКГ, Rg органов грудной полости (для исключения тимомы, гемосидероза), костей черепа, запястья. Трансфузиологический анамнез.

Анемия Фанкони Ø Ø Ø Ø Ø Впервые описана в 1927 г под названием детского емейного апластического миелоза. Тип наследования: аутосомно – рецессивный, высокийуровень хромосомных обераций. Патогенез: в основе эритроцитопении лежит продукция дефектных клеток, длительность жизни которых резко укорочена, костный мозг не компенсирует повышенное разрушение эритроцитов. В гибели эритроцитов играют роль как внутриклеточные (врожденные энзимопении), так и внеклеточные факторы. Одновременно вовлекаются и другие ростки кроветворения, в том числе и миелоидный ряд, что указывает на поражение стволовой клетки Клиника: чаще встречается у мальчиков. Дебют 4 -12 лет * пренатальная гипотрофия (масса ≤ 2500 ги рост 45 -48 см) * в дальнейшем отставание в физическом развитии сохраняется (недостаточная продукция СТГ, ТТГ, ГТГ, АКТГ) * отставание костного возраста на 2 -5 лет Типичны аномалии развития: - микроцефалия, гидроцефалия; - микрофтальмия, косоглазие, эпикант, гипертелоризм, атрофия зрительного нерва; - аплазия или гипоплазия большого пальца кисти и метакарпальной кости, лучекостевой синостоз, косорукость, синдактилия, гипоплазия тазобедренных суставов

- ВПС Ø - врожденные аномалии мочевых путей и почек Ø - снижение слуха Ø -бронзовокоричневая пигментация (за счет отложения меланина в клетках базальног слоя эпидермиса) диффузная, усиливающаяся в местах естественных складок пятна «кофе с молоком» . Ø Ребенок с рождения бледный, снижен апетит, малоподвижный, позднее отмечается головная боль, слабость, снижение толерантности к физнагрузкам, имеются изменения ЦНС в виде замкнутости, «психической инфантильности» . Участи детей отмечается тахикардия, пониженное АД, приглушение сердечных тонов, расширение границ сердечной тупости преимущественно влево. Печень. Селезенка не увеличены. Классификация: I тип – классический с выраженными пороками развития II тип – с малыми аномалиями развития, гиперпигментацией кожи Ø Особенности гемопоэза: развивается нарастающая пангемоцитопения: снижение эритроциов до 0. 66 -0. 94*109/л, гемоглобин 30 -65 г/л. Анемия нормохромная или гиперхромная, анизоцитоз, умеренный пойкилоцитоз. Ретикулоциты 2 -2. 5% по мере прогрессирования ретикулоцитоз снижается. Лейкопения стойкаядостигает максимума в терминальном периоде (гранулоциты составляют до 0. 1*109/л)

Ø Ø Ø Тромбоцитопения достигает значительной степени до единичных тромбоцитов в мазке. СОЭ повышена 30 -80 мм/ч. Имеется стресс-эритропоэз, который характеризуется макроцитозом. Высокий уровень Hb Fдо 15%-45%( N 2%), высокий уровень эритропоэтина, наличием i-антигена. Стернальный пунктат нормо- или гипоклеточный. Количество в пределах нормы. Увеличено количество клетокэритроидного росткас задержкой их созревания, анизоцитоз, базофильной пунктацией в нормобластах иногда появляются мегалобласты. Гранулоцитарный росток, мегакариоцитарный рост Разрастание жировой ткани Течение: хроническое с чередованием ремиссий и обострений. Гипоплазия костного мозга постепенно прогрессирует. Без лечения через 2 года после диагностики умирают 80% больных, а через 4100%.
. Ø Метод выбора в лечении анемии Фанкони.")
Лечение Ø 1. Трансплантация костного мозга (ТКМ). Ø Метод выбора в лечении анемии Фанкони. Проводится ТКМ от HLAидентичного сиблинга с использованием смягченного кондиционирования – торакоабдоминального облучения в дозе 6 Гр и циклофосфамида в дозе 20 мг/кг. Такой подход позволяет излечить около 70 -75% больных. Ø 2. Консервативное лечение андрогенами Ø При отсутствии донора для ТКМ назначают лечение стероидными анаболиками в течении 3 -6 мес. Улучшение наступает через 6 -8 недель (увеличивается количество ретикулоцитов и гемоглобина, а затем лейкоцитов, число тромбоцитов не повышается длительно). Ответ на терапию андрогеами имеет прогностическое значение: при ответе средняя выживаемость 9 лет, без ответа 2, 5 глда. Ø 3. Заместительная гемотрансфузионная терапия Ø Показания: уровень гемоглобина меньше 80 г/л , абсолютное число нейтрофилов меньше 1, 0*109/л, число тромбоцитов меньше 20*109/л. Ø 4. Гемопоэтические ростовые факторы Ø Могут быть назначены в качестве пробной терапии при неэффективности обычного лечения и отсутствии совместимого донора. Используют Г-КСФ, ГМ-КСФ. Ø 5. Попытки генной терапии.

АНЕМИЯ БЛЕКФЕНА-ДАЙМОНДА Частота 1: 1000000 живых новорожденных, мальчики и девочки заболевают одинаково часто. Наследование: подавляющее большинство (75%) – спорадические случаи, возможно аутосомно-доминантное, аутосомнорециссивное или сцепленное с X-хромасомой. Клиника: масса тела и рост нормальные. Психомоторное развитие нормальное. Врожденные пороки развития встречаются реже (25%) чем при анемии Фанкони. Характерен фенотип: - волосы цвета пакли, - курносый нос, - большая верхняя губа, - гипертелоризм. Кожа и слизистые бледные с первых дней жизни по мере прогрессирования приобретает восковидный оттенок, к 5 -6 годам серый (гемосидероз). Гепатомегалия, спленомегалия. Костный возраст отстает от паспортного на 4 -5 лет, запаздывает смена молочных зубов.
. Гемоглобин и эритроциты снижаются")
Особенности гемопоэза: анемия нормохромная, гипорегираторная или арегинераторная (ретикулоцитопения 0 -1%). Гемоглобин и эритроциты снижаются до черезвычайно низких цифр. Лейкоциты и тромбоциты на нормальном уровне, при длительном течении тромбоцитопения и нейтропения. СОЭ увеличено – 20 -80 мм/ч. Миелограмма: костный мозг - нормоклеточный, по мере прогресирования - гипоклеточность. Эритроедный росток резко сужен (диагностический критерий – эритробластов менее 5% ядросодержащих клеток). Миелоидный и мегакариоцитарный ростки не изменены. Число ретикулярных клеток и лимфоцитов не увеличено, плазматических клеток не изменено. Биохимия: высокий уровень эритроцитарной аденозиндезаминазы, Hb. F в N, повышенное содержание i-антигена в эритроцитах, увеличено содержание эритропоэтина в сыворотке. Лечение: у 75% больных эффективным средством являются кортикостероиды в сочетании с анболическими стероидами. Гормоны оказывают влияние на пролиферацию косто-мозговых клеток, кортикостероиды приводят к увеличению ретикулоцитов, уровню гемоглобина, возможно восстановление эритропоэза.

Наиболее эффективен преднизолон 1 -1, 5 мг/кг в сутки. Через 2 нед при положительном эффекте лечение продолжают снижая дозу до поддерживающей – ½, 1/3, от лечебной в течение полутора месяцев. Из анаболических стероидов – метиландростенолон 1 -2 мг/кг в сутки. Осложнения терапии: гепатит, вирилизация, прибавка в массе тела, задержка Na в организме. Проводимое лечение позволяет детям дожить до 8 -15 лет. Причиной смерти может быть цирроз печени, острый гепатит, гемосидероз органов.

Приобретенные апластические анемии Ø Начинаются в возрасте 14 -15 лет. Основным проявлением являются симптомы угнетения нормального кроветворения, гипоксией тканей и органов. Острая АА Внезапное начало с высокой гектической лихорадки, резкой слабости, рвотой, болями в животе, геморрагиями на коже и слизистых различной величины и локализации, выраженная бледность. Быстро присоединяется некротическая ангина. За 5 -7 дней развивается тяжелая панцитопения, увеличивается печень. Увеличение сердца в поперечнике, тахикардия, систолический шум на верхушке, одышка. Адинамия, анорексия, кровотечения( ДВС-синдром I-II фаза). Отсутствие ремиссии или стабилизации процесса не смотря на интенсивное лечение. Летальный исход через 4 -8 недель. Подстрая АА Развивается менее бурно, часто предшевствуют прививки, температура нормальная или субфебрильная, геморагииполиморфны, часты носовые и кровотечения в ЖКТ, сетчатку, различные отделы уха. Предвестниками кровоизлияний в мозг, появление геморрагий на верхней половине туловища, лице. Тахикардия, расширение границ сердца, систолический шум на верхушке, Снижение АД. Умеренное увеличение печени

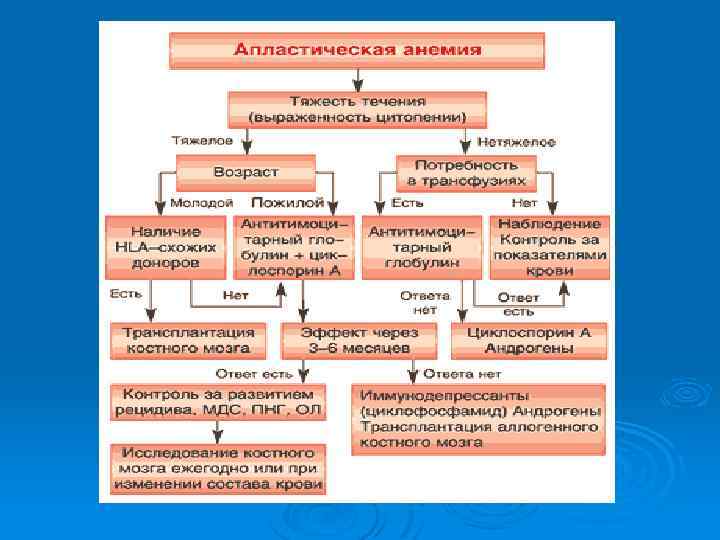
Хроническая АА Слабость утомляемость, гиперпигментация отдельных участков кожи в подмышечных областях. Симптомы кровоточивости в виде точечных кровоилияний, экхимозов, носовых кровотечений, Температура нормальная, Выраженная бледность , прозрачные мочки, бескровные слизистые Лечение 1. 2. 3. 4. 5. Трансплантация костного мозга Иммуносупрессия (Антилимфоцитарный глобулин, циклоспорин, высокие дозы метилпреднизолона, циклофосфана) Гемопоэтические ростковые факторы Андрогены Симптоматическая терапия (гемокомпонентная, антибактериальная, гемостатическая)
Апл анемии08.ppt