405e8cc785cb67b8b66c9fc4192dec8e.ppt
- Количество слайдов: 64



“ACCREDITATION FOR MICROBIOLOGICAL LABORATORIES – General requirements” Aliki Stathopoulou Kyiv, 5 -6 February 2015 TAIEX Workshop on the Accreditation of Medical and Clinical Laboratories (55902)

CONTENTS 1. INTRODUCTION 2. PERSONNEL 3. ACCOMMODATION AND ENVIRONMENTAL CONDITIONS 4. VERIFICATION AND VALIDATION OF TEST METHODS 5. ESTIMATION OF UNCERTAINTY 6. EQUIPMENT 7. REAGENTS AND CULTURE MEDIA 8. REFERENCE MATERIALS AND REFERENCE CULTURES 9. PRE-EXAMINATION PROCESSES 10. QUALITY CONTROL 11. TEST REPORTS

1. INTRODUCTION

Microbiology Laboratory Services The services offered by a clinical microbiology laboratory, are including: 1. Analysis of clinical samples of patients with infections 2. Information on the correct choice of antimicrobial treatment 3. Recording and surveillance of antimicrobial resistance in the hospital 4. Update the principles for outbreaks of infections that threaten public health Microbiological testing is taken to include detection, isolation, enumeration and identification of micro-organisms (viruses, bacteria, fungi) and their metabolites in samples derived from the human body, by using either classical or molecular microbiology methods (as confirmation/ identification steps, or as alternative detection methods).

Microbiology Laboratory Services A Clinical Microbiology Laboratory, in order to ensure the quality of its results, so that they are accurate, reliable and reproducible, should implement a quality system applied in the overall operation, commonly accepted and officially recognized. Proper implementation and evaluation of such a quality system are covered by accreditation. Accreditation ensures the technical competence of the laboratory for specific tests (scope of accreditation), and the operation of a Quality Management System in accordance with the requirements of EN ISO 15189: 2012.

2. PERSONNEL

Laboratory Director • Laboratory should be supervised by an experienced person, qualified to degree level in medical microbiology or equivalent. • Alternative qualifications may meet requirements where a staff member has extensive relevant experience relating to the laboratory's scope of accreditation. • Laboratory director shall ensure that there appropriate numbers of staff to provide services that meet the needs and requirements of the users.

Laboratory Personnel • Staff should be sufficient trained and have relevant practical work experience before being allowed to perform work covered by the scope of accreditation without supervision, or before being considered as experienced for supervision of accredited work. • Ongoing competence in relation to the results of internal and external quality control, should be monitored objectively with provision for retraining where necessary. • The laboratory shall use personnel who are employed by, or under contract to, the laboratory. • The laboratory shall have job descriptions that describe responsibilities, authorities and tasks for all personnel.

Laboratory Personnel • A comprehensive vaccination policy should be formulated for all personnel taking into consideration work activities, rather than job title. • Informed consent, preferably in writing, should be obtained before screening and vaccination. The provision of information about the relevant vaccine-preventable diseases should be provided as part of the process of informed consent. If recommended vaccines are declined, obtain signed documentation using a declination form. This may be requested by the employer to meet their Occupational Health and Safety requirements. Employers should take all reasonable steps to encourage non-immune workers to be vaccinated.

Laboratory Personnel • An up-dated register should be kept by a designated staff member, containing details of staff vaccine preventable disease history, vaccination, antibody and test results, record of vaccines consented/refused or non-response to vaccination. • Recommended vaccines are Tetanus / Diphtheria, Hepatitis B, Influenza. • Work practices should include the use of standard and additional precautions to minimise exposure to blood and body fluids. If exposure does occur, guidelines for post exposure prophylaxis should be followed. Ensure that post exposure guidelines are easily accessible 24 hours a day. • The laboratory supervisor must ensure that laboratory personnel receive appropriate training regarding their duties, the necessary precautions to prevent exposures, and exposure evaluation procedures. Personnel must receive annual updates or additional training when procedural or policy changes occur.

3. ACCOMMODATION AND ENVIRONMENTAL CONDITIONS

Premises • The typical laboratory is comprised of the testing facilities (where specific microbiological testing and associated activities are carried out) and ancillary facilities (entrances, corridors, administration blocks, cloakrooms and toilets, storage rooms, archives, etc. ). • Space should be sufficient to allow work areas to be kept clean and tidy. The space required should be commensurate with the volume of analyses handled and the overall internal organisation of the laboratory. The space should meet the requirements of national regulations when available. • Workrooms should be appropriately ventilated and at a suitable temperature. It is important that natural ventilation should be avoided in clean rooms or workrooms handling pathogens. • Access to the laboratory area is strongly restricted and visits of external persons shall be documented.

Premises • It is generally considered as good practice to have separate locations, or clearly designated areas, for at least the following: Øsample receipt and storage area; Øsample preparation; Øexamination of samples, including incubation; Ømaintenance of reference organisms; Østorage of culture media and reagents; Ømedia and equipment preparation, including sterilization; Østerility assessment; Ødecontamination; Øcleaning of glassware and other equipment;

Premises • For a molecular microbiology laboratory the following four distinct working areas are strongly recommended for: a) preparation of reagents b) sample handling (e. g. splitting and extraction) c) setting up of reactions d) amplification and analysis of amplicons. Dedicated pipettes, tips, centrifuges, tubes, adequate protective clothing, vials, heating blocks etc. should be located in each work area. • If rooms with different pressures are available, always the highest positive pressure should prevail in the area of nucleic acid extraction and set up of reaction mixtures. • DNA amplification should be conducted in a dedicated section of the laboratory in a positive pressurised room.

Contamination control • Any microbiological specimen requiring nucleic acid-based testing shall arrive as straight as possible without previous manipulation and shall be opened exclusively in the PCR sample handling area. • Where materials are used for multiple types of testing, separate collection of samples is preferable for splitting. Any splitting shall be performed immediately in the PCR sample handling area only. • Where molecular techniques are undertaken, monitoring for DNA contaminants should be undertaken by using a “No Template Control” (NTC).

Contamination control • The laboratory should have arrangements to minimise risks of cross-contamination, where these are significant to the type of test being performed. The ways to achieve these objectives are, for example: Øto construct the laboratory according to the 'no way back’ layout principle; Øto carry out procedures in a sequential manner using appropriate precautions to ensure test and sample integrity Øto segregate activities by time or space.

Contamination control • Reduction of contamination may be achieved by having: Øsmooth surfaces on walls, ceilings, floors and benches (the smoothness of a surface is judged on how easily it may be cleaned). Tiles are not recommended as bench covering material; Øconcave joints between the floor, walls and ceiling; Øno furniture, documents or other items other than those strictly necessary for testing activities; Øminimal opening of windows and doors while tests are being carried out; Øsun shades placed on the outside; Øfluid conveying pipes not passing above work surfaces unless placed in hermetically sealed casings; (continued on next page)

Contamination control Øa dust-filtered air inlet for the ventilation system; Øseparate hand-washing arrangements, preferably non manually controlled; Øwhere air conditioners are used, filters should be appropriate, inspected, maintained and replaced according to the type of work being carried out; Øcupboards up to the ceiling; Øno rough and bare wood; Øwooden surfaces of fixtures and fittings adequately sealed; Østored items and equipment arranged to facilitate easy cleaning;

Biosafety • Each laboratory should develop or adopt a biosafety or operations manual that identifies the hazards that will or may be encountered, and that specifies practices and procedures designed to minimize or eliminate exposures to these hazards. • Risk assessment is an important responsibility for directors and principal investigators of microbiological and biomedical laboratories. It is recommended that the laboratory does a Risk assessment for the appropriate selection of microbiological practices, safety equipment, and facility safeguards that can prevent laboratory-associated infections (LAI).

Biosafety For the development of this process, the international Laboratory Biorisk Management Standard CWA 15793: 2008 could be used, in combination with commercially available software programs, which provide methods and tools for visualization of the relative risks, and identification of risk mitigation measures. Examples of topics covered: ØBiorisk Management Policy ØHazard Identification, risk assessment, and risk control ØRoles, responsibilities, and authorities ØTraining, awareness and competence ØOperational control ØEmergency response and contingency plans ØInventory monitoring and control ØAccident and incident investigation ØInspection and audit ØBiorisk management review

Personnel safety • Personal protective clothing appropriate to the type of testing being performed (including, if necessary, gloves, coats, gowns, shoe covers, boots, respirators, face shields, safety glasses, or goggles, etc. ) should be worn in the microbiological laboratory and removed before leaving the area. • This is particularly important in the molecular biology laboratory, where for example, movement from an area of high DNA load to one of low DNA load may unwittingly introduce cross -contamination. A change of the laboratory coat may suffice when moving between areas. • Adequate hand washing facilities should be available and a policy regarding appropriate glove use, should be in place to avoid the spreading of micro-organisms in the laboratory.

Environmental monitoring • The laboratory shall monitor, control and record environmental conditions while special attention shall be paid to factors such as temperature, sterility, dust, lighting, hazardous fumes, humidity, sound and vibration levels (especially regarding the work areas used for microscopy examination). • An appropriate environmental monitoring programme shall be devised, including, for example, frequent use of air settlement plates for bacterial and fungal contaminants as well as periodic surface swabbing for a variety of relevant micro-organisms, especially pathogens. Acceptable background counts should be assigned and there should be a documented procedure for dealing with situations in which these limits are exceeded. Analysis of data should enable trends in levels of contamination to be determined.

Hygiene • A documented program for regular cleaning and disinfection, of surfaces and equipment, shall be applied to the laboratory (i. e. with 5% sodium hypochloride or 70% alcohol, UV lamps) taking into account the results of environmental monitoring and the possibility of cross-contamination. There should be a procedure for dealing with spillages. • Decontamination of all cultures, stocks, and other potentially infectious materials, should be done before disposal, using an effective method.

4. VERIFICATION AND VALIDATION OF TEST METHODS

Selection of methods • The laboratory shall use appropriate test methods to meet the specific needs in each case. The methods could be: Østandard methods (CE IVD approved), which require verification by the Laboratory Østandard methods used outside their intended scope or modifidied standard methods, which require analytical validation and clinical verification Ølaboratory-designed/developed methods (in-house methods), which require analytical validation and clinical verification

Selection of methods • The validation of microbiological test methods should reflect actual test conditions. This may be achieved by using naturally contaminated products or products spiked with a predetermined level of contaminating organisms. • The analyst should be aware that the addition of contaminating organisms to a matrix only mimics in a superficial way the presence of the naturally occurring contaminants. However, it is often the best and only solution available. • The extent of validation necessary will depend on the method and the application. The laboratory shall validate standard methods applied to matrices not specified in the standard procedure.

Selection of methods • The methods used should be appropriate for the tests carried out in the laboratory, according to the requirements of clinical evaluation (community patients, hospital patients, etc. ), which are indicated by the manufacturers of analyzers and / or rely on methods established by international scientific organizations. • Beyond the initial confirmation of the correct application of the method, at least once per year, re-evaluation of the efficiency of laboratories to perform specific methods correctly is performed. The annual review of the Laboratory’s efficiency is conducted through analysis and systematic evaluation of all data from internal and external quality schemes.

Quantitative and semi-quantitative tests • Verification Trueness: Assessment of trueness is achieved by using reference materials and results of participation in external quality assurance schemes. Repeatability: In order to check repeatability at measurements per organism are required, depending on the difficulty and the inherent danger of the microorganism and the substrate in two to three levels of inoculated samples. The standard deviation and coefficient of variation (CVr%) of measurements is estimated. (continued on next page)

Quantitative and semi-quantitative tests Intermediate precision / reproducibility: ØFirst method: Use of two to three levels of inoculated samples (spiked samples). At least ten measurements are made for each sample at different dates (if the sample is stable) or with different operators or different reagents. ØSecond method: From quality control charts (control charts) over a one year period. ØThird method: From factorial experimental design of multiple measurements of the sample in different experimental conditions. One - way ANOVA application may be used for calculating the SDR. The precision of a method is satisfactory if the relative standard deviation (CV%) is less than 10% or less than the permissible limit specified by the relevant literature. (continued on next page)
: The detection limit is determined by")
Quantitative and semi-quantitative tests Limit of Detection (LOD): The detection limit is determined by examining artificially contaminated samples, which are the product of successive decimal dilutions of the tested microorganism. The initial number of cfu should be known. The samples are analyzed by laboratory analysts under reproducibility conditions and the smallest number of microorganisms that can be detected is the detection limit of the method. The number of samples to be analyzed depends on the microorganism and the substrates with a justification for the number by the laboratory.

Qualitative tests • Verification Repeatability / Intermediate precision / laboratory reproducibility: Analysis of a number of positive and negative samples per microorganism and substrate is required from the laboratory analysts. The number of samples to be analyzed depends on microorganisms and substrates with a justification for the numbers from the laboratory. Sensitivity and Specificity : The sensitivity is calculated from the ratio: true positive / total positive results and the specificity from the ratio: true negative results / total negative results. (continued on next page)
: The detection limit of the method is determined")
Qualitative tests Limit of Detection (LOD): The detection limit of the method is determined by examining artificially contaminated samples, which are the product of successive decimal dilutions of the microorganism examined. The samples are analyzed by the laboratory analysts, under conditions of intralaboratory reproducibility. The smallest number of microorganisms that can be detected is the detection limit of the method. The number of samples to be analyzed depends on the microorganism and the substrates with a justification of the number by the laboratory. The percentage of "positive" results should not deviate from the approved rate of sensitivity of the method, which is derived from the validation studies of the method or the relevant literature.
 tests • Validation In-house methodologies are validated the same way CE-IVD")
Laboratory-developed internal (in-house) tests • Validation In-house methodologies are validated the same way CE-IVD methodologies are validated by their manufacturers. In the case of accreditation according to ISO 15189, in addition to analytical validation, clinical validation is also required (diagnostic sensitivity and specificity, NPV, PPV, clinical utility). For Research-use-only tests the laboratory may be accredited according to ISO 17025, without the need for clinical validation.
 tests • Requirements regarding the validation of in-house laboratory developed methods")
Laboratory-developed internal (in-house) tests • Requirements regarding the validation of in-house laboratory developed methods are the following: Trueness: Assessment of trueness is achieved either by using control samples commercially provided or reference materials, results from proficiency testing schemes and recovery experiments. Furthermore, trueness is checked by: ØMethod comparison: it is required to compare the method with a CE-IVD Kit (or when absent, with another commercial kit or other established methodology) by analysing a number of samples (e. g. 20 negative and 20 positive in qualitative parameters, 10 negative and 30 positive with a broad range of values in quantitative parameters) and appropriate statistical analysis of the results. ØIn multiparametric methodologies where more than one microorganism is detected, the laboratory must have a complete panel of control samples in order to validate the accuracy of the method used. (continued on next page)
 tests Repeatability/ Intermediate precision/intralaboratory reproducibility: At least ten (10) measurements of")
Laboratory-developed internal (in-house) tests Repeatability/ Intermediate precision/intralaboratory reproducibility: At least ten (10) measurements of control samples in at least two levels (negative / weakly positive, 2 -3 x LOD) on different days and using different lots of reagents. The CV% of the measurements is calculated (it can be drawn from control charts within a period of 3 or 6 months). Limit of Detection (LOD): Validation of LOD is calculated by applying Probit Analysis at five concentration levels of the reference material, around the LOD. The verification of LOD should be done with samples at -20% of LOD and +20% of LOD. For multiparametric methodologies, the process should be repeated for each detected parameter. Limit of Quantification (LOQ) in quantitative methods: the point where a satisfactory CV% (e. g. 20%) is achieved. (continued on next page)
 tests Linearity and Measuring Range for quantitative methods: For an in-house")
Laboratory-developed internal (in-house) tests Linearity and Measuring Range for quantitative methods: For an in-house Q-PCR methodology able to measure in the range of 10 log units, using at least 7 points (in triplicate) by diluting appropriate reference material is necessary for obtaining the standard curve (n = 5) and performing linearity check (dilutions shall cover at least 5 log units). Analytical Specificity: for molecular microbiological methods check with electrophoresis and DNA Sequencing the obtained PCR product (and Tm in Q-PCR) for the presence of genetically-similar organisms or organisms that are often found in the samples analyzed in the laboratory.

5. ESTIMATION OF UNCERTAINTY

Quantitative and semi-quantitative tests • The estimation of uncertainty budget is based on reproducibility data and on elements of systematic errors (bias), as determined by the results of proficiency testing schemes (at least ten samples is required). • The desired homogeneity of the sample should be a specific element of evaluation, depending on the biological material, in relation with the type and number of microorganisms it contains. A comprehensible protocol for sample size and processing (suitable / unsuitable sample) should be used for this parameter.
 (real or spiked)")
Quantitative and semi-quantitative tests • Analysis of 10 different samples (i) (real or spiked) at various levels, in both conditions of reproducibility A and B (e. g. two analysts, different reagent lots etc) Measurement of the number of microorganisms/ cfu. Xi. A and Xi. B Calculating log 10 (Xi. A) and log 10 (Xi. B) Expanded uncertainty U = 2 x SR (log 10)

6. EQUIPMENT

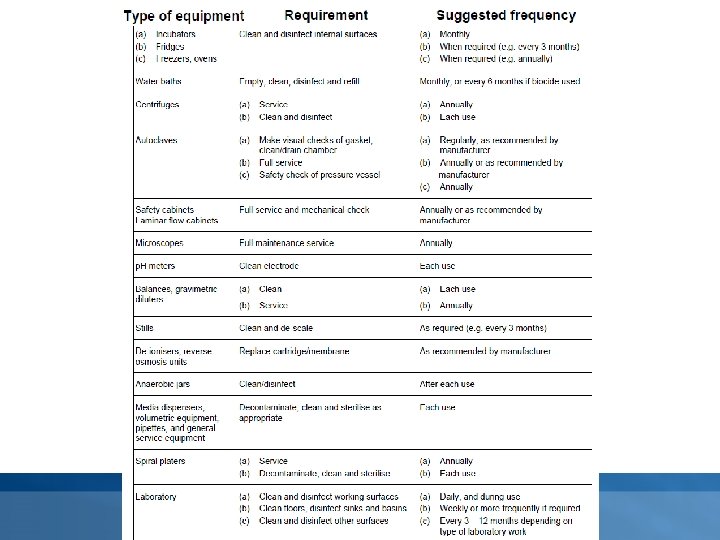
Maintenance • Maintenance of essential equipment shall be carried out at specified intervals as determined by factors such as the frequency of use. Detailed records shall be kept. • Typically, the following items of equipment will be maintained by cleaning and servicing, inspecting for damage, by general verification of suitability and, where relevant, sterilising: Øgeneral service equipment – filtration apparatus, glass or plastic containers (bottles, test tubes), glass or plastic Petri dishes, sampling instruments, wires or loops (platinum, nickel/chromium or disposable plastic); Øwater baths, incubators, microbiological cabinets (laminar flow and safety cabinets), autoclaves, homogenisers, fridges, freezers; Øvolumetric equipment – pipettes, automatic dispensers, spiral platers; Ømeasuring instruments – thermometers, timers, balances, p. H meters, colony counters.
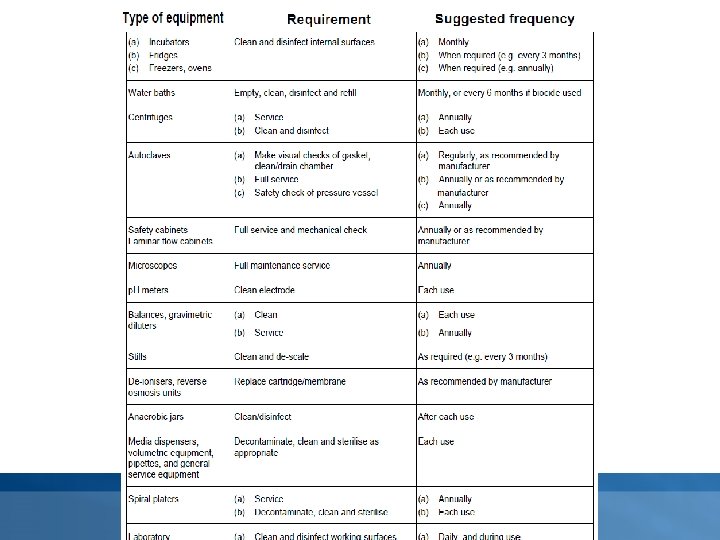

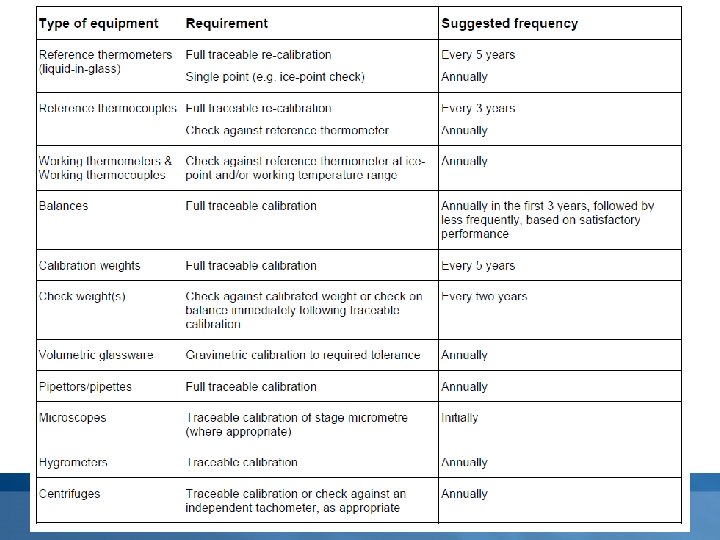
Calibration and performance verification • The laboratory must establish a programme for the calibration and performance verification of equipment which has a direct influence on the test results. • The frequency of such calibration and performance verification will be determined by documented experience, being based on Øneed/usage (i. e. incubation, decontamination, sterilization, storage, monitoring) ØType (i. e. temperature measurement devices, incubators, water baths, ovens, autoclaves, thermal cyclers) Øprevious performance (i. e. efficiency, effectiveness, number of equipment failures and down time)

7. REAGENTS AND CULTURE MEDIA

Reagents - Media • Laboratories should ensure that the quality of reagents used is appropriate for the test concerned. They should verify the suitability of each batch of reagents critical for the test, initially and during its shelf-life, using positive and negative control organisms which are traceable to recognised national or international culture collections. • In-house prepared and Ready-to-use media : The suitable performance of culture media, diluents and other suspension fluids should be checked, where relevant, with regard to: Ørecovery or survival maintenance of target organisms; Øinhibition or suppression of non-target organisms; Øbiochemical (differential and diagnostic) properties; Øphysical properties (e. g. color, thickness, bubbles, surface smoothness, hemolysis)

Reagents - Media Laboratories should set acceptability criteria for incoming batches of media appropriate for its testing needs, taking into consideration the validation exercise, and at least the following: Øname of the media and list of components, including any supplements; Øshelf-life and the acceptability criteria applied; Østorage conditions; Østerility check; Øcheck of growth of target and non-target control organisms used (with their culture collection references) and acceptability criteria; Øphysical checks and the acceptability criteria applied; Ødate of issue of specification.
, media,")
Reagents - Media • Laboratories shall ensure that all reagents (including stock solutions), media, diluents, and other suspending fluids are adequately labelled to indicate, as appropriate: Øidentity Øconcentration Østorage conditions Ødate of opening Øpreparation date Øvalidated expiry date and/or recommended storage periods. • The person responsible for preparation should be identifiable from records.

8. REFERENCE MATERIALS AND REFERENCE CULTURES
 and certified reference materials (CRM) provide essential traceability")
Reference materials • Reference materials (RM) and certified reference materials (CRM) provide essential traceability in measurements and are used, for example: Øto Øto demonstrate the accuracy of results; calibrate equipment; monitor laboratory performance; validate methods; enable comparison of methods; demonstrate quality of culture media; demonstrate consistent performance of kits. • If possible, reference materials should be used in appropriate matrices.

Reference cultures • Traceable reference cultures are required for establishing acceptable performance of media (including test kits), for validating methods and for assessing/evaluating on-going performance. • Reference strains may be sub-cultured to provide reference stocks. Master Culture storage (-70° or -20° C) 1 st generation Long term storage Stock cultures (multiple) Storage (-20° C) 2 nd generation Storage from 6 months to 5 years Working culture Storage (-4° C in non-selective media) 3 rd generation Storage for 4 weeks

9. PRE-EXAMINATION PROCESSES

Instructions for sampling • Collection instructions should contain at least the following: ØThe appropriate sampling site or sample type for each test ØThe appropriate time (i. e. before the receipt of antimicrobial drugs) ØThe adequate amount of the primary sample for each test according specific instructions ØThe appropriate tube/specimen container ØThe appropriate medium for the sample transportation and sample conservation (additives) ØInstructions for the appropriate labeling of the container ØInstructions for sample transportation (i. e. appropriate temperature intervals, appropriate time frame, safety issues)

Test request • Request form for testing should provide: ØPatient identification ØName of the ordering physician or other person authorized to order testing ØClinician’s address ØType of primary sample collected ØAnatomic site where appropriate ØTest requested ØPatient gender ØDate of birth ØPertinent clinical information as appropriate for purposes of test interpretation ØDate and time sample collection and receipt in the laboratory ØPreferred sample type (venous, arterial, capillary, urine, spinal fluid) ØType of anticoagulant ØSample volume considered acceptable

Sample handling • Criteria used to determine if a specimen is unacceptable for testing : ØSpecimen is received without a requisition ØRequisition is received without a specimen ØRequisition or specimen label lacks two patient identifiers ØRequisition or specimen label information is illegible ØRequisition and specimen label information is not identical ØRequisition and/or specimen mislabeled (Patient identifiers inaccurate) ØIncorrect specimen container/tube is used ØDate of collection is not recorded ØTime of collection is not recorded (continued on next page)

Sample handling ØSpecimen is clotted ØSpecimen is too old for testing ØSpecimen container is leaking ØSpecimen quantity is insufficient ØSpecimen contamination, dilution or other interfering substances affect specimen integrity; example: hemolysed, lipemic ØInappropriate specimen ØDuplicate test request • Specimens not meeting the requirements may be accepted by the laboratory under the following circumstances: ØSpecimen type identified as irretrievable ØSpecimen has been acquired through an invasive procedure

10. QUALITY CONTROL

Internal Quality Control • A programme of periodic checks is necessary to demonstrate that variability (i. e. between analysts and between equipment or materials etc. ) is under control. All tests included in the laboratory’s scope of accreditation need to be covered. • The programme is determined according to the type of examination, the frequency of execution and the daily program of the Laboratory and may involve: Øthe use of spiked samples with variable contamination levels Øthe use of spikes/naturally contaminated samples Øthe use of reference materials (including proficiency testing scheme test materials) Øreplicate testing Ørandom rescreening: The laboratory staff reviews a representative number of samples, proportional to the laboratory workload (measurement of inter-observative and intra-observative agreement)

Diameter Levey-Jennings curve Day

External Quality Control • Participation should be annually or more frequent, in accordance with the program of the provider of the proficiency testing scheme and for all tests, in: Øan approved interlaboratory program (Proficiency Testing providers like INSTAND, NEQAS, LABQUALITY) or Øan interlaboratory comparison scheme, whose provider complies with specific requirements and is evaluated based on these requirements by participants

11. TEST REPORTS

Reporting of results • Test results are reported on forms approved by the laboratory management under the Quality System and clearly identify the following: ØPatient ØDate and time of specimen collection ØTest performed ØReference or normal range ØThe laboratory interpretation where appropriate ØName or initial of person performing the test ØAuthorized signature of person reviewing the report and releasing the results • The results are eligible without transcription mistakes and are reported only to persons authorized to receive them, such as the ordering physician or nursing staff in a hospital environment. The report must also indicate if the sample received was unacceptable for testing. Reports of test results are quality records and are kept for a period of time, as specified by the laboratory or a government requirement.

Thank you!
405e8cc785cb67b8b66c9fc4192dec8e.ppt