L5.ppt
- Количество слайдов: 29



або ВРОДЖЕНІ ПОМИЛКИ МЕТАБОЛІЗМУ

Метаболізмсукпність взаємоповязаних біохімічних процесів в організмі Кожна реакція каталізується ферментом

Механізм: А В ЕА накопич. С e. В D ЕС недостат. Ідентифіковано > 500 захворювань і класифіковано як inborn errors of metabolism вроджені помилки метаболізму

Класифікація за типом пошкодженого метаболічного шляху Підкласи: • Аміноацидопатії Органічні ацидурії Дефекти цикла сечовини Дефекти дихального ланцюга мітохондрій Глікогенози Дефекти мітохондрійного окисненя Пероксисомні заворювання 31% 27% 21% 12% 8% 8% 4%
 2. Х – зчеплені 3. Мітохондрійні")
Типи успадкування 1. Аутосомно - рецесивні (переважна більшість) 2. Х – зчеплені 3. Мітохондрійні 4. Аутосомно – домінантні (зрідка) Характер прояву мутації Мутації в структурній частині гена • < активності фермента; • > активності фермента (рідко); • порушення стабільності фермента; • відсутність фермента (рідко); • поліморфізм ферментів. Мутації в регуляторних частинах гена зміна транскрипції гена.
 1: 8 000 Хвороба Тея-Сакса 1:")
Частота Аутосомно-рецесивний тип успадкування Фенілкетонурія (сер. по Європі) 1: 8 000 Хвороба Тея-Сакса 1: 120 000 (серед євреїв-ашкеназі) 1: 3 000 Хвороба Гоше (накопичення глюкоцереброзидів в лізосомах клітин ) 1: 40 000 Х-зчеплений рецесивний тип успадкування Х-зчеплена адренолейкодистрофія Мукополісахаридоз тип II 1: 40 000 1: 70 000

Клінічні ознаки метаболічних порушень -Нейротоксичний вплив – затримка розвитку, згодом судоми -Можуть виявлятись у різному віці, найчастіше у ранньому дитинстві Діагностика Часто симптоми не специфічні Необхідні лабораторні методи діагностики, а не клінічні - Пренатальна діагностика при плануванні наступної дитини - Для деяких хвороб можлива часткова або повна клінічна корекція

Методи діагностики біохімічні, молекулярно-генетичні Ген Білок ДНК-диіагностика Ензимодіагностика та методи аналізу білків Метаболіти Хроматографічні або інші кількісні методи

Тандемна масспетрометрія -Одочасний аналіз багатьох метаболітів - швидкий - достатньо краплі крові 100 контроль Pro Внутрішні стандарти % Intensity Ala Phe 50 Leu + Ile Phe Leu Tyr Gln Val Ala Gly 140 Ser 160 Val 180 Tyr Glu Cit Met 200 220 m/z, amu Asp 240 Glu 260 280 300
 та")
Хвороба кленового сиропу накопичення амінокислот з розгалуженим вуглецевим ланцюгом (лейцину, ізолейцину і валіну) та токсичних продуктів їхнього метаболізму 100 Leu + Ile % Intensity Внутрішні стандарти 50 Val Phe Pro Gln Ala Gly 140 Ala Ser 160 Val 180 Leu Phe Cit Met 200 220 m/z, amu Tyr Asp 240 Glu 260 280 300

Тирозинемія 100 Tyr Внутрішні стандарти % Intensity Pro 50 Ala Phe Leu + Ile Gln Gly 140 Ala Val Ser 160 Val 180 Leu Cit Met 200 220 m/z, amu Tyr Asp 240 Glu 260 280 300
 • 1934 р. А. Фелінг описав, • 1940 р.")
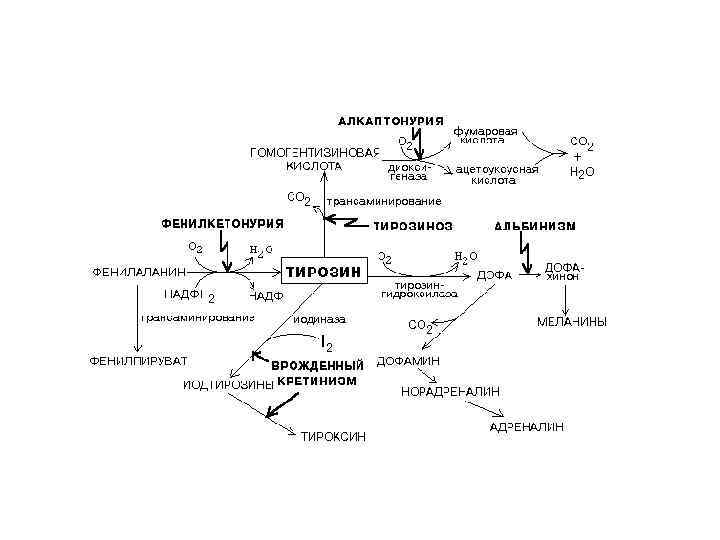
ПОРУШЕННЯ ОБМІНУ АМІНОКИСЛОТ ФЕНІЛКЕТОНУРІЯ (ФКУ) • 1934 р. А. Фелінг описав, • 1940 р. Л. Пенрозо – ФКУ, • 1947 р. Ербіс – недостатність феніл-аланін-4 -гідроксилази, • з 60 рр. скринінг новонароджених, • 1985 р. Лідскі і сп. - ген РАН – 12 q 22 -24. 1 • 1986 р. РАН секвенували: перша мутація IVS 12 nt 1, • 1987 p. - найпоширеніша мутація R 408 W
 12 q 22 -24. 1 к.")
ГЕН РАН БІЛОК Pah PAH – PKU (фенілкетонурія) 12 q 22 -24. 1 к. ДНК 2448 пн: 13 екз. - 90 тпн: 57 пн (9) – 895 пн (13); 12 інтр. - 85 тпн: 1000 пн (7)- 23, 5 тпн (3). Pah – фенілаланін-4 -гідроксилаза синтезується в гепатоцитах 52 k. D: 451 AK - гомотетрамер: 3 домени – регуляторний, каталітичний, тетрамеризаційний Успадкування: а-р Гомологія: 90% з РАН кролика, 92% з РАН миші;
 - затримка розвитку, - розумова відсталість помірна, - епілетичні напади, агресивність,")
Фенотип: (без лікування) - затримка розвитку, - розумова відсталість помірна, - епілетичні напади, агресивність, шкірні зміни, - недостатність пігментації, - “мишачий запах” (фенілоцтова к-та)

МЕТАБОЛІЗМ ФЕНІЛАЛАНІНУ PH – фенілаланінгідроксилаза, ВН 2 –дигідроптеридин, ВН 4 – тетрагідроптеридин

Частота фенілкетонурії у різних популяціях • • • Японія – 1/125 000 Фінляндія – 1/ 100 000 Китай – 1/ 17 000 Італія – 1/ 17 000 Великобританія – 1/14 000 США (білі) – 1/10 000 Україна – 1/8 000 Шотландія – 1/5 000 Туреччина – 1/2 600
 – 895")
к. ДНК 2448 пн: 13 екз. – 90 тпн: 57 пн (9) – 895 пн (13); 12 інтр. – 85 тпн: 1000 пн (7) – 23, 5 тпн (3). Мутації в гені РАН РКU

MУТАЦІЇ ГЕНА РАН більше 400 в Європі переважають 29 Мутації Локаліз. Тип Амінок-та R 408 W R 158 Q R 252 W IVS 12 nt 1 IVS 10 nt 546 12 E 5 E 7 E 12 інтp. 10 інтр. C G C С G T A T А A арг три R 413 P R 408 Q Y 414 C 12 E 12 E C A A G G G арг тир Частота Укр три глі арг “ “ 57% 40% Ірландія 3, 7% 2, 95% 1% 13% Данія 1, 5% 30% Єгипет про 0, 5% глу цис 1, 6% 30% Японія

Успішна терапія ФКУ – дієта з різким зменшенням фенілаланіну до 16 -18 років

ПОРУШЕННЯ ФЕРМЕНТІВ РЕПАРАЦІЇ • Варіанти пігментної ксеродерми • Синдроми спонтанної хромосомної нестабільності • Синдроми перечасного старіння
 Гени: XPA - XPG (7 груп комплементації)")
Варіанти пігментної ксеродерми Пігментна ксеродерма (Xeroderma pigmentosum) Гени: XPA - XPG (7 груп комплементації) Білки беруть участь у реплікації пошкодженої ДНК на лідерному ланцюзі (3’ 5’) ХРА – 9 q 22. 3 кодує Zn 2+ finger protein – розпізнання фотопродуктів Ген XPG 13 q 32 кодує специфічну ендонуклеазу (3'-нуклеаза при ексцизійній репарації); Гени ХРВ і ХРD кодують субодиниці TFIIH (Transcription factor II H) Успадкування: а-р Фенотип: - гіперчутливість до світла, - ненормальна пігментація, - схильнсть до раку шкіри, - дефекти шкіри.
 Сockayne syndrome Гени: CSB (ERCC 6) 10 q 11. 23 “excision")
Синдром Коккейна (CS) Сockayne syndrome Гени: CSB (ERCC 6) 10 q 11. 23 “excision repair cross-complementing, complementation group 6. ” СSA (ERCC 8) – 5 q 12 Білок порушення репарації ДНК В культурі клітин синтез РНК пригнічується УФ світлом Частота: 1: 250 тис. нн Фенотип: розумова відсталість, світлочутливість, мікроцефалія, глибоко посаджені очі, глухота, нейрональна дисфунуція, недостатність росту, катаракта…ТЖ ~ 6 -7 р. Успадкування: а-р
 Ген BLM - 15 q")
Синдроми спонтанної хромосомної нестабільності – с. Блума (Bloom syndrome) Ген BLM - 15 q 26. 1, Білок Blm – ДНК-геліказа Фенотип: хромосомна нестабільність (40%), cхильність до онкогенезу (остеосаркоми), імунна недостатність, лицеві еритеми, чутливість до УФ, гіпо- і гіперпігментація шкіри. Успадкування: а-р (євреї Ашкеназі) Обміни між сестринськими хроматидами

Анемія Фанконі вроджений дефект кісткового мозку Гени: FANC F-G, FANC – 16 q 24. 3 Білки: участь в репарації ДНК Частота: 1 : 350 тис (частіше у - євреїв ашкеназі -1/30 000 - Південна Африка, іспанські цигани – 1/40 000 Фенотип: розумова відсталість не значна, скелетні дефекти, ушкодження кісткового мозку, порушення пігментації, лейкоз Успадкування : а-р Хромосомні розриви
 Телеектазії Ген: Atm -11 q 22. 3 Білок: АТМ кіназа (вкорочення")
АТАКСІЯ-ТЕЛЕАНГІЕКТАЗІЯ (с. Луї-Бар) Телеектазії Ген: Atm -11 q 22. 3 Білок: АТМ кіназа (вкорочення теломер, надактивність p 53) Частота: 1: 50 тис. нн Фенотип: -мозочкова атаксія, -нейродегенерація, -імунна недостатність, -телеангіектазії на склері, - надчутливість до рентген. випром, -схильність до онкогенезу, -розумова відсталість. ТЖ ~ 20 р. Успадкування: а-р 7 q/14 p перебудови

Синроми передчасного старіння с. Хатчінсона-Гілфорда- прогерія дітей Ген: LMNA -1 q 21. 2 Білок ламіни А (компонент ядерної ламіни) Частота: 1: 1 млн Фенотип: нездатність до росту, вади кісток, плямиста гіпопігментація шкіри, відсутність росту волосся, дзьобоподібний ніс, утворення атеросклеротичних бляшок, ТЖ – 13 - 20 р. Успадкування: de novo

с. Вернера прогерія дорослих Ген: WRN – 6 p 25. 2, Білок: геліказа Rec. Q 1432 ак Частота: 1 : 200 тис (Японія 1 : 40 тис) Фенотип: симптоми старіння після 16 -20 років, ТЖ – 30 -48 р. Успадкування: А-Р

L5.ppt