

6cc19e35fa4a9f6cabf0972a58de36e4.ppt
- Количество слайдов: 77
 4. DIFFUSION
4. DIFFUSION
 4. 1 DEFINITION Spreading out, mixing. The diffusion of gases and liquids refers to their mixing without an external force. Diffusion is defined as a process of mass transfer of individual molecules of a substance, brought about by random molecular motion and associated with a concentration gradient.
4. 1 DEFINITION Spreading out, mixing. The diffusion of gases and liquids refers to their mixing without an external force. Diffusion is defined as a process of mass transfer of individual molecules of a substance, brought about by random molecular motion and associated with a concentration gradient.
. Through") 4. 2. MECHANISMS Diffusion could occur: Throughout a single bulk phase (solution’s homogeneity). Through a barrier, usually a polymeric membrane (drug release from film coated dosage forms, permeation and distribution of drug molecules in living tissues, passage of water vapor and gases through plastic container walls and caps, ultrafiltration).
4. 2. MECHANISMS Diffusion could occur: Throughout a single bulk phase (solution’s homogeneity). Through a barrier, usually a polymeric membrane (drug release from film coated dosage forms, permeation and distribution of drug molecules in living tissues, passage of water vapor and gases through plastic container walls and caps, ultrafiltration).
 4. 2. MECHANISMS Diffusion through a barrier may occur through either: Simple molecular permeation (molecular diffusion) through a nonporous membrane. This process depends on the dissolution of the permeating molecules in the barrier membrane. Movement through pores and channels which involves the passage of the permeating molecules through solvent filled pores in the membrane rather than through the polymeric matrix itself.
4. 2. MECHANISMS Diffusion through a barrier may occur through either: Simple molecular permeation (molecular diffusion) through a nonporous membrane. This process depends on the dissolution of the permeating molecules in the barrier membrane. Movement through pores and channels which involves the passage of the permeating molecules through solvent filled pores in the membrane rather than through the polymeric matrix itself.
 and a") 4. 2. MECHANISMS A B Diffusion through a homogenous nonporous membrane (A) and a porous membrane with solvent (usually water) filled pores (B)
4. 2. MECHANISMS A B Diffusion through a homogenous nonporous membrane (A) and a porous membrane with solvent (usually water) filled pores (B)
 4. 2. MECHANISMS Both mechanisms usually exist in any system and contribute to the overall mass transfer or diffusion. Pore size, molecular size and solubility of the permeating molecules in the membrane polymeric matrix determine the relative contribution of each of the two mechanisms.
4. 2. MECHANISMS Both mechanisms usually exist in any system and contribute to the overall mass transfer or diffusion. Pore size, molecular size and solubility of the permeating molecules in the membrane polymeric matrix determine the relative contribution of each of the two mechanisms.
 4. 2. MECHANISMS Passage of steroidal molecules substituted with hydrophilic groups in topical preparation through human skin involves transport through the skin appendages (hair follicles, sebum ducts and sweat pores in the epidermis) as well as molecular diffusion through the stratum corneum.
4. 2. MECHANISMS Passage of steroidal molecules substituted with hydrophilic groups in topical preparation through human skin involves transport through the skin appendages (hair follicles, sebum ducts and sweat pores in the epidermis) as well as molecular diffusion through the stratum corneum.
 4. 2. MECHANISMS By combining the two mechanisms for diffusion through a membrane we can achieve a better representation of a membrane on the molecular scale. A membrane can be visualized as a matted arrangement of polymer strands with branching and intersecting channels.
4. 2. MECHANISMS By combining the two mechanisms for diffusion through a membrane we can achieve a better representation of a membrane on the molecular scale. A membrane can be visualized as a matted arrangement of polymer strands with branching and intersecting channels.
 4. 2. MECHANISMS An illustration of the microstructure of a cellulose membrane used in the filtration processes showing the intertwining nature of fibers and aqueous channels
4. 2. MECHANISMS An illustration of the microstructure of a cellulose membrane used in the filtration processes showing the intertwining nature of fibers and aqueous channels
 4. 2. MECHANISMS Depending on the size and shape of the diffusing molecules, they may pass through the tortuous pores formed by the overlapping strands of the polymer. The other alternative is to dissolve in the polymer matrix and pass through the film by simple diffusion.
4. 2. MECHANISMS Depending on the size and shape of the diffusing molecules, they may pass through the tortuous pores formed by the overlapping strands of the polymer. The other alternative is to dissolve in the polymer matrix and pass through the film by simple diffusion.
 4. 4. FICK’S FIRST LAW OF DIFFUSION Flux J is the amount, M, of material flowing through a unit cross section, S, of a barrier in unit time, t. J = d. M / S. dt The units of the Flux J are g. cm-2. sec-1.
4. 4. FICK’S FIRST LAW OF DIFFUSION Flux J is the amount, M, of material flowing through a unit cross section, S, of a barrier in unit time, t. J = d. M / S. dt The units of the Flux J are g. cm-2. sec-1.
 4. 4. FICK’S FIRST LAW OF DIFFUSION According to the diffusion definition, the flow of material is proportional to the concentration gradient. Concentration gradient represents a change of concentration with a change in location. Concentration gradient is referred to as dc/dx where c is the concentration in g/cm 3 and x is the distance in cm of movement perpendicular to the surface of the barrier ( i. e. across the barrier).
4. 4. FICK’S FIRST LAW OF DIFFUSION According to the diffusion definition, the flow of material is proportional to the concentration gradient. Concentration gradient represents a change of concentration with a change in location. Concentration gradient is referred to as dc/dx where c is the concentration in g/cm 3 and x is the distance in cm of movement perpendicular to the surface of the barrier ( i. e. across the barrier).
 Fick’s First") 4. 4. FICK’S FIRST LAW OF DIFFUSION J dc/dx J = -D*(dc/dx) Fick’s First Law In which D is the diffusion coefficient of the permeating molecule (diffusant, penetrant) in cm 2/sec. The negative sign in the equation signifies that the diffusion occurs in the direction of decreased concentration. Flux J is always a positive quantity (dc/dx is always negative)
4. 4. FICK’S FIRST LAW OF DIFFUSION J dc/dx J = -D*(dc/dx) Fick’s First Law In which D is the diffusion coefficient of the permeating molecule (diffusant, penetrant) in cm 2/sec. The negative sign in the equation signifies that the diffusion occurs in the direction of decreased concentration. Flux J is always a positive quantity (dc/dx is always negative)
 4. 4. FICK’S FIRST LAW OF DIFFUSION D is more correctly referred to as Diffusion Coefficient rather than constant since it does not ordinarily remain constant and may change with concentration. Diffusion will stop when the concentration gradient no longer exists (i. e. , when dc/dx = 0).
4. 4. FICK’S FIRST LAW OF DIFFUSION D is more correctly referred to as Diffusion Coefficient rather than constant since it does not ordinarily remain constant and may change with concentration. Diffusion will stop when the concentration gradient no longer exists (i. e. , when dc/dx = 0).
 4. 5. FICK’S SECOND LAW OF DIFFUSION Fick’s first law examined the mass diffusion across a unit area of a barrier in a unit time. Fick’s second law examines the rate of change of diffusant concentration with time at a point in the system. The diffusant concentration c in a particular volume element changes only as a result of a net flow of diffusing molecules into or out of the specific volume unit.
4. 5. FICK’S SECOND LAW OF DIFFUSION Fick’s first law examined the mass diffusion across a unit area of a barrier in a unit time. Fick’s second law examines the rate of change of diffusant concentration with time at a point in the system. The diffusant concentration c in a particular volume element changes only as a result of a net flow of diffusing molecules into or out of the specific volume unit.
 4. 5. FICK’S SECOND LAW OF DIFFUSION Input C Output Volume element Bulk medium
4. 5. FICK’S SECOND LAW OF DIFFUSION Input C Output Volume element Bulk medium
 4. 5. FICK’S SECOND LAW OF DIFFUSION A difference in concentration results from a difference in input and output. The rate of change in the concentration of the diffusant with time in the volume element ( c/ t) equals the rate of change of the flux (amount diffusing) with distance ( J/ x) in the x direction. dc/dt = d. J/dx
4. 5. FICK’S SECOND LAW OF DIFFUSION A difference in concentration results from a difference in input and output. The rate of change in the concentration of the diffusant with time in the volume element ( c/ t) equals the rate of change of the flux (amount diffusing) with distance ( J/ x) in the x direction. dc/dt = d. J/dx
") 4. 5. FICK’S SECOND LAW OF DIFFUSION dc/dt = d. J/dx J = -D*(dc/dx) Differentiating with respect to x d. J/dx = D*(d 2 c/dx 2) substituting dc/dt from the top equation dc/dt = D*(d 2 c/dx 2)
4. 5. FICK’S SECOND LAW OF DIFFUSION dc/dt = d. J/dx J = -D*(dc/dx) Differentiating with respect to x d. J/dx = D*(d 2 c/dx 2) substituting dc/dt from the top equation dc/dt = D*(d 2 c/dx 2)
 This") 4. 5. FICK’S SECOND LAW OF DIFFUSION dc/dt = D*(d 2 c/dx 2) This equation represents diffusion in the x axis only. In order to describe diffusion in three dimensional space, Fick’s second law can be written as dc/dt = D*(d 2 c/dx 2 + d 2 c/dy 2 + d 2 c/dz 2 )
4. 5. FICK’S SECOND LAW OF DIFFUSION dc/dt = D*(d 2 c/dx 2) This equation represents diffusion in the x axis only. In order to describe diffusion in three dimensional space, Fick’s second law can be written as dc/dt = D*(d 2 c/dx 2 + d 2 c/dy 2 + d 2 c/dz 2 )
 4. 5. Fick’s Second Law of Diffusion Input C Output Volume element Bulk medium
4. 5. Fick’s Second Law of Diffusion Input C Output Volume element Bulk medium
 4. 6. STEADY STATE DIFFUSION An important condition in diffusion is that of steady state Fick’s first law gives the flux (or rate of diffusion throughout unit area) in the steady state of flow. Fick’s second law refers in general to a change in concentration of diffusant with time, at any distance, x (i. e. , a nonsteady state of flow). Steady state may be described, however, in terms of the second law.
4. 6. STEADY STATE DIFFUSION An important condition in diffusion is that of steady state Fick’s first law gives the flux (or rate of diffusion throughout unit area) in the steady state of flow. Fick’s second law refers in general to a change in concentration of diffusant with time, at any distance, x (i. e. , a nonsteady state of flow). Steady state may be described, however, in terms of the second law.
 4. 6. Steady state Diffusion n Diffusion Cells: n In a diffusion cell, two compartments are separated by a polymeric membrane. n The diffusant is dissolved in a proper solvent and placed in one compartment while the solvent alone is placed in the other. n The solution compartment is described as Donor Compartment because it is the source of the diffusant in the system while the solvent compartment is described as the Receptor Compartment.
4. 6. Steady state Diffusion n Diffusion Cells: n In a diffusion cell, two compartments are separated by a polymeric membrane. n The diffusant is dissolved in a proper solvent and placed in one compartment while the solvent alone is placed in the other. n The solution compartment is described as Donor Compartment because it is the source of the diffusant in the system while the solvent compartment is described as the Receptor Compartment.
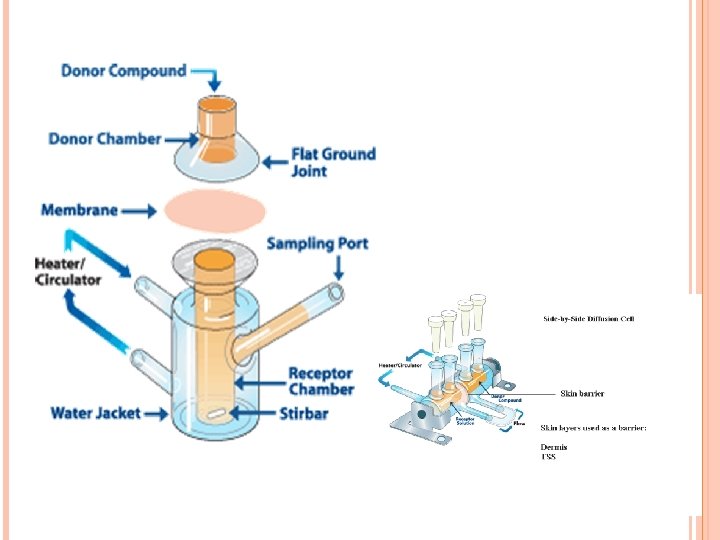
 4. 6. STEADY STATE DIFFUSION In diffusion experiments, the solution in the receptor compartment is constantly removed and replaced with a fresh solvent to keep the concentration of the diffusant passing from the donor compartment at a low level. This is referred to as the Sink Condition.
4. 6. STEADY STATE DIFFUSION In diffusion experiments, the solution in the receptor compartment is constantly removed and replaced with a fresh solvent to keep the concentration of the diffusant passing from the donor compartment at a low level. This is referred to as the Sink Condition.
 4. 6. STEADY STATE DIFFUSION Donor Compartment Receptor Compartment Pure solvent Diffusant Solution Flux in Me m b r a n e (c) Flux out Flow of solvent to maintain sink condition
4. 6. STEADY STATE DIFFUSION Donor Compartment Receptor Compartment Pure solvent Diffusant Solution Flux in Me m b r a n e (c) Flux out Flow of solvent to maintain sink condition
 4. 6. STEADY STATE DIFFUSION As the diffusant passes through the membrane from the donor compartment (d) to the receptor compartment (r), the concentration in the donor compartment (Cd) will fall while the concentration in the receptor (Cr) will rise. However, the concentration in the receptor compartment is always maintained at very low levels because of the sink condition. This means that Cr << Cd
4. 6. STEADY STATE DIFFUSION As the diffusant passes through the membrane from the donor compartment (d) to the receptor compartment (r), the concentration in the donor compartment (Cd) will fall while the concentration in the receptor (Cr) will rise. However, the concentration in the receptor compartment is always maintained at very low levels because of the sink condition. This means that Cr << Cd
 4. 6. STEADY STATE DIFFUSION When the system has been in existence for a sufficient period of time (determined by the nature of barrier and the rate of removal of the diffusant by the sink system), the rate of change in concentration in the two compartments with time will become constant. However, the concentration in the two compartments is not the same.
4. 6. STEADY STATE DIFFUSION When the system has been in existence for a sufficient period of time (determined by the nature of barrier and the rate of removal of the diffusant by the sink system), the rate of change in concentration in the two compartments with time will become constant. However, the concentration in the two compartments is not the same.
 = 0 Since") 4. 6. STEADY STATE DIFFUSION dc/dt = D*(d 2 c/dx 2) = 0 Since D is not equal to (0), then d 2 c/dx 2 should be 0. Since d 2 c/dx 2 is a second derivative, and is equal to (0) the first derivative dc/dx should be a constant. This means that the concentration gradient dc/dx across the membrane is constant (linear relationship between concentration c and distance or membrane thickness h)
4. 6. STEADY STATE DIFFUSION dc/dt = D*(d 2 c/dx 2) = 0 Since D is not equal to (0), then d 2 c/dx 2 should be 0. Since d 2 c/dx 2 is a second derivative, and is equal to (0) the first derivative dc/dx should be a constant. This means that the concentration gradient dc/dx across the membrane is constant (linear relationship between concentration c and distance or membrane thickness h)
 4. 6. STEADY STATE DIFFUSION Receptor Compartment Donor Compartment C 1 Cd High concentration of diffusant molecules C 2 Cr 0 Thickness of barrier h
4. 6. STEADY STATE DIFFUSION Receptor Compartment Donor Compartment C 1 Cd High concentration of diffusant molecules C 2 Cr 0 Thickness of barrier h
, Fick’s first law may") 4. 6. STEADY STATE DIFFUSION In such systems (diffusion cells), Fick’s first law may be written as: J = d. M / S. dt = D *(C 1 -C 2)/h C 1 and C 2 are the concentrations within the membrane and are not easily measured. However they can be calculated using the partition coefficient (K)and the concentrations on the donor (Cd) and receptor (Cr) sides which can be easily measured
4. 6. STEADY STATE DIFFUSION In such systems (diffusion cells), Fick’s first law may be written as: J = d. M / S. dt = D *(C 1 -C 2)/h C 1 and C 2 are the concentrations within the membrane and are not easily measured. However they can be calculated using the partition coefficient (K)and the concentrations on the donor (Cd) and receptor (Cr) sides which can be easily measured
 4. 6. STEADY STATE DIFFUSION K = C 1/Cd = C 2/Cr Replacing C 1 and C 2 with KCd and KCr d. M / S. dt = D (C 1 -C 2)/h = D(K Cd -K Cr)/h d. M / S. dt = DK(Cd - Cr)/h d. M / dt = DSK(Cd - Cr)/h
4. 6. STEADY STATE DIFFUSION K = C 1/Cd = C 2/Cr Replacing C 1 and C 2 with KCd and KCr d. M / S. dt = D (C 1 -C 2)/h = D(K Cd -K Cr)/h d. M / S. dt = DK(Cd - Cr)/h d. M / dt = DSK(Cd - Cr)/h
 4. 6. STEADY STATE DIFFUSION If the sink condition holds in the receptor compartment Cd>>Cr 0 and Cr drops out of the equation which becomes d. M / dt = DSKCd /h The term DK/h is referred to as the Permeability Coefficient or Permeability (P) and has the units of linear velocity (cm/sec). The equation simplifies further to become d. M / dt = PSCd
4. 6. STEADY STATE DIFFUSION If the sink condition holds in the receptor compartment Cd>>Cr 0 and Cr drops out of the equation which becomes d. M / dt = DSKCd /h The term DK/h is referred to as the Permeability Coefficient or Permeability (P) and has the units of linear velocity (cm/sec). The equation simplifies further to become d. M / dt = PSCd
 4. 6. STEADY STATE DIFFUSION Permeability can be calculated from experimental data obtained from diffusion cells. - If Cd remains relatively constant throughout time, P can be obtained from the slope of a linear plot of M versus t. M=PS Cd t
4. 6. STEADY STATE DIFFUSION Permeability can be calculated from experimental data obtained from diffusion cells. - If Cd remains relatively constant throughout time, P can be obtained from the slope of a linear plot of M versus t. M=PS Cd t
 4. 6. STEADY STATE DIFFUSION - If Cd changes appreciably with time, then P can be obtained from the slope of log Cd versus t. log Cd = log Cd(0) - (PSt/2. 303 Vd)
4. 6. STEADY STATE DIFFUSION - If Cd changes appreciably with time, then P can be obtained from the slope of log Cd versus t. log Cd = log Cd(0) - (PSt/2. 303 Vd)
 Steady state A m o u n t D i ffu s e d Nonsteady state Lag time t. L Time
Steady state A m o u n t D i ffu s e d Nonsteady state Lag time t. L Time
 4. 7. CONTROLLED RELEASE COATED TABLETS Dissolution Polymeric membrane Diffusion Solid Core Saturated solution in equilibrium with solid core Unit Activity
4. 7. CONTROLLED RELEASE COATED TABLETS Dissolution Polymeric membrane Diffusion Solid Core Saturated solution in equilibrium with solid core Unit Activity
 4. 7. CONTROLLED RELEASE COATED TABLETS If the excess solid in the dosage form is depleted, the activity ( ) decreases as the drug diffuses out of the system and the release rate falls exponentially (First order release). A constant activity dosage form may not exhibit a zero order release in the initial stages (membrane hydration, dissolution of part of the solid core to create the saturated solution) and a lag time may be observed.
4. 7. CONTROLLED RELEASE COATED TABLETS If the excess solid in the dosage form is depleted, the activity ( ) decreases as the drug diffuses out of the system and the release rate falls exponentially (First order release). A constant activity dosage form may not exhibit a zero order release in the initial stages (membrane hydration, dissolution of part of the solid core to create the saturated solution) and a lag time may be observed.
 4. 7. CONTROLLED RELEASE COATED TABLETS The straight line in the last figure can be presented by the following equation: M = SDKCd(t-t. L)/h and the lag time is given by: t. L = h 2/6 D When diffusivity can be calculated, presuming a knowledge of the membrane thickness, h Also, knowing P, the thickness h can be calculated from t. L = h/6 P
4. 7. CONTROLLED RELEASE COATED TABLETS The straight line in the last figure can be presented by the following equation: M = SDKCd(t-t. L)/h and the lag time is given by: t. L = h 2/6 D When diffusivity can be calculated, presuming a knowledge of the membrane thickness, h Also, knowing P, the thickness h can be calculated from t. L = h/6 P
 5. 7. PERCUTANEOUS ABSORPTION OF DRUGS:
5. 7. PERCUTANEOUS ABSORPTION OF DRUGS:
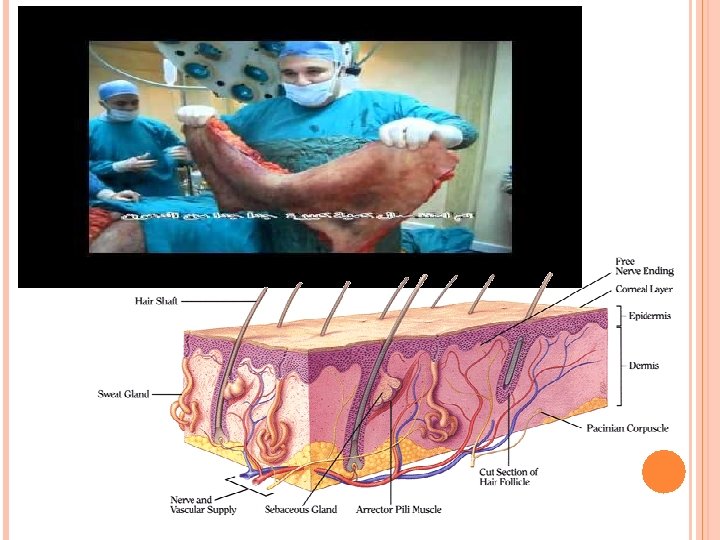
 5. 7. PERCUTANEOUS ABSORPTION OF DRUGS: Percutanous absorption include: Transcellular diffusion Diffusion through channels between the cells Diffusion through sebaceous ducts Transfollicular diffusion Diffusion through sweat ducts
5. 7. PERCUTANEOUS ABSORPTION OF DRUGS: Percutanous absorption include: Transcellular diffusion Diffusion through channels between the cells Diffusion through sebaceous ducts Transfollicular diffusion Diffusion through sweat ducts
 5. 7. PERCUTANEOUS ABSORPTION OF DRUGS: It is an oversimplification to assume that one route prevails under all conditions. Yet after steady state conditions have been established, transcellular diffusion through the stratum corneum most likely predominates. In the early stages of drug penetration, diffusion through skin appendages ( hair follicles, sebaceous and sweat ducts) may be significant. These “shunt” pathways are even important in the steady state diffusion in the case of large polar molecules e. g. some polar corticosteroids.
5. 7. PERCUTANEOUS ABSORPTION OF DRUGS: It is an oversimplification to assume that one route prevails under all conditions. Yet after steady state conditions have been established, transcellular diffusion through the stratum corneum most likely predominates. In the early stages of drug penetration, diffusion through skin appendages ( hair follicles, sebaceous and sweat ducts) may be significant. These “shunt” pathways are even important in the steady state diffusion in the case of large polar molecules e. g. some polar corticosteroids.
 5. 7. PERCUTANEOUS ABSORPTION OF DRUGS: The factors influencing the penetration of a drug into the skin include: The concentration of the dissolved drug Cs. The partition coefficient Kvs between the skin and the vehicle, which is a measure of the relative affinity of the drug for skin and vehicle. Diffusion coefficients, which represent resistance to drug molecule movement through vehicle (Dv) and skin (Ds).
5. 7. PERCUTANEOUS ABSORPTION OF DRUGS: The factors influencing the penetration of a drug into the skin include: The concentration of the dissolved drug Cs. The partition coefficient Kvs between the skin and the vehicle, which is a measure of the relative affinity of the drug for skin and vehicle. Diffusion coefficients, which represent resistance to drug molecule movement through vehicle (Dv) and skin (Ds).
 5. 7. PERCUTANEOUS ABSORPTION OF DRUGS: The relative magnitude of the two diffusion coefficients, Dv and Ds, determines whether release from vehicle or passage through the skin is the rate -limiting step.
5. 7. PERCUTANEOUS ABSORPTION OF DRUGS: The relative magnitude of the two diffusion coefficients, Dv and Ds, determines whether release from vehicle or passage through the skin is the rate -limiting step.
 OCCLUSION VS. SKIN BARRIER FUNCTION REF. : SKIN RESEARCH AND TECHNOLOGY 2002: 8: 1– 6 OCCLUSION: Refers to skin covered directly or indirectly by impermeable films or substances such as tape, gloves, textiles garments, wound dressings etc. In addition, certain topical vehicles that contain fats and/or polymers oils (petrolatum, paraffin, etc. ) may also generate occlusive effects. Occlusion, because of its simplicity, is widely utilized to enhance the penetration of applied drugs in clinical practice. However, occlusion does not increase percutaneous absorption of all chemicals. It may increase penetration of lipid-soluble, nonpolar molecules but has less effect on polar molecules: a trend of occlusion-induced absorption enhancement with increasing penetrant lipophilicity is apparent. In practice, increasing skin penetration rates of applied drug is far from simple.
OCCLUSION VS. SKIN BARRIER FUNCTION REF. : SKIN RESEARCH AND TECHNOLOGY 2002: 8: 1– 6 OCCLUSION: Refers to skin covered directly or indirectly by impermeable films or substances such as tape, gloves, textiles garments, wound dressings etc. In addition, certain topical vehicles that contain fats and/or polymers oils (petrolatum, paraffin, etc. ) may also generate occlusive effects. Occlusion, because of its simplicity, is widely utilized to enhance the penetration of applied drugs in clinical practice. However, occlusion does not increase percutaneous absorption of all chemicals. It may increase penetration of lipid-soluble, nonpolar molecules but has less effect on polar molecules: a trend of occlusion-induced absorption enhancement with increasing penetrant lipophilicity is apparent. In practice, increasing skin penetration rates of applied drug is far from simple.
 Skin barrier function can be ascribed to the macroscopic structure of the stratum corneum, consisting of alternating lipoidal and hydrophylic regions. For this reason, physicochemical characteristics of the chemical, such as partition coefficient, structure, and molecular weight, play an important role in determining the facility of absorption. Another factor to consider in drug percutaneous absorption is the vehicle in which the drug is formulated, as it influences drug release from the formulation. In addition, the anatomical site may influence the effects of occlusion on percutaneous absorption. In many industrial and food fields, protective gloves or clothing are required to protect the workers from hazardous materials or to maintain hygiene. In turn, these protective measures may produce negative events due to the nature of occlusion, which often causes stratum corneum hyperhydration and reduces the protective barrier properties of the skin. Many gloves do not resist the penetration of low molecular weight chemicals: those chemicals may enter the glove and become trapped on the skin under occlusion for many hours, possibly leading to irritation, and more seriously, to dermatitis or eczematous changes.
Skin barrier function can be ascribed to the macroscopic structure of the stratum corneum, consisting of alternating lipoidal and hydrophylic regions. For this reason, physicochemical characteristics of the chemical, such as partition coefficient, structure, and molecular weight, play an important role in determining the facility of absorption. Another factor to consider in drug percutaneous absorption is the vehicle in which the drug is formulated, as it influences drug release from the formulation. In addition, the anatomical site may influence the effects of occlusion on percutaneous absorption. In many industrial and food fields, protective gloves or clothing are required to protect the workers from hazardous materials or to maintain hygiene. In turn, these protective measures may produce negative events due to the nature of occlusion, which often causes stratum corneum hyperhydration and reduces the protective barrier properties of the skin. Many gloves do not resist the penetration of low molecular weight chemicals: those chemicals may enter the glove and become trapped on the skin under occlusion for many hours, possibly leading to irritation, and more seriously, to dermatitis or eczematous changes.
 Wound dressings have been employed to speed the healing processes in acute and chronic wounds by keeping healing tissues moist and increasing superficial wound epithelialization. However, occlusive or semiocclusive dressings can increase microorganisms and hence induce wound infections. A significant increase in the density of Staphylococcus aureus and lipophilic diphtheroids were observed after 24 h occlusion in eczematous and psoriatic skin. The effects of occlusion on skin are complex, and may produce profound changes including altering epidermal lipids, DNA synthesis, epidermal turnover, p. H, epidermal morphology, sweat glands, Langerhans cells stresses, etc. Evaluation and investigation of the impact of occlusion on barrier function are important in the fields of skin physiology, pathology, and pharmacology, and dermatology.
Wound dressings have been employed to speed the healing processes in acute and chronic wounds by keeping healing tissues moist and increasing superficial wound epithelialization. However, occlusive or semiocclusive dressings can increase microorganisms and hence induce wound infections. A significant increase in the density of Staphylococcus aureus and lipophilic diphtheroids were observed after 24 h occlusion in eczematous and psoriatic skin. The effects of occlusion on skin are complex, and may produce profound changes including altering epidermal lipids, DNA synthesis, epidermal turnover, p. H, epidermal morphology, sweat glands, Langerhans cells stresses, etc. Evaluation and investigation of the impact of occlusion on barrier function are important in the fields of skin physiology, pathology, and pharmacology, and dermatology.
 has been well recognized as the principle") Skin Barrier Function Stratum corneum (horny layer) has been well recognized as the principle barrier of the skin. It is a cellular tissue, a fabric of cornified cells, creating a tough, flexible, coherent membrane which acts as a two–way barrier, by minimizing water loss, electrolytes, and other body constituents and decreasing the entry of noxious substances from the external environment. Topical application is an obvious choice in the treatment of dermatological disease, but pharmaceutical agents applied topically have also been shown to enter the systemic circulation. Physical, chemical, and pathological factors can disturb barrier function: even changes in environmental humidity may induce pathophysiologic alterations. Maintenance of the stratum corneum structural integrity is critical to barrier function. Increasing stratum corneum hydration can progressively reduce its barrier efficiency. Stratum corneum is extremely hygroscopic: it can pick up 500% of its dry weight in less than 1 h following immersing in water, swelling vertically to 4– 5 times its original width.
Skin Barrier Function Stratum corneum (horny layer) has been well recognized as the principle barrier of the skin. It is a cellular tissue, a fabric of cornified cells, creating a tough, flexible, coherent membrane which acts as a two–way barrier, by minimizing water loss, electrolytes, and other body constituents and decreasing the entry of noxious substances from the external environment. Topical application is an obvious choice in the treatment of dermatological disease, but pharmaceutical agents applied topically have also been shown to enter the systemic circulation. Physical, chemical, and pathological factors can disturb barrier function: even changes in environmental humidity may induce pathophysiologic alterations. Maintenance of the stratum corneum structural integrity is critical to barrier function. Increasing stratum corneum hydration can progressively reduce its barrier efficiency. Stratum corneum is extremely hygroscopic: it can pick up 500% of its dry weight in less than 1 h following immersing in water, swelling vertically to 4– 5 times its original width.
 Effects of Occlusion on Barrier Function Healthy stratum corneum typically has a water content of 10– 20%. Occlusion can block diffusional water loss from skin surface, thereby increasing stratum corneum hydration, swelling corneocytes and promoting water uptake into intercellular lipid domains. Water content can be increased up to 50% with occlusion: even short time (30 min) exposure can result in significantly increased stratum corneum hydration. With 24 h occlusion, the relative water content in stratum corneum can be increased significantly. 24 h occlusion can induce morphological changes on the surface, deepening skin furrows. Water under occlusion may disrupt barrier lipids and damage the stratum corneum in a way similar to surfactants. Kligman studied hydration dermatitis in man: one week with an impermeable plastic film did not injure skin, 2 weeks was moderately harmful to some but not all subjects, 3 weeks consistently induced dermatitis.
Effects of Occlusion on Barrier Function Healthy stratum corneum typically has a water content of 10– 20%. Occlusion can block diffusional water loss from skin surface, thereby increasing stratum corneum hydration, swelling corneocytes and promoting water uptake into intercellular lipid domains. Water content can be increased up to 50% with occlusion: even short time (30 min) exposure can result in significantly increased stratum corneum hydration. With 24 h occlusion, the relative water content in stratum corneum can be increased significantly. 24 h occlusion can induce morphological changes on the surface, deepening skin furrows. Water under occlusion may disrupt barrier lipids and damage the stratum corneum in a way similar to surfactants. Kligman studied hydration dermatitis in man: one week with an impermeable plastic film did not injure skin, 2 weeks was moderately harmful to some but not all subjects, 3 weeks consistently induced dermatitis.
 Optimal Occlusion Obviously, occlusion alone may damage skin barrier function. Application of chemicals/drugs under occlusion can increase penetration of chemicals and antigens into the skin and therefore can increase dermatitis. Local reactions (i. e. irritation and/or sensitization) of TDDS, typically occlusive patches placed on the skin surface for 1– 7 days to deliver the drugs into the systemic circulation, have been widely reported. However, reactions can be minimized with immunosuppressive agents, antioxidants, local anaesthetics, and other anti-irritant technologies. Advancements in design and construction of protective garments and wound dressings may reduce the level of skin hydration and dermatitis. Application of optimal hydrocolloid patches that absorb water in both liquid and vapor form can also decrease irritation. A natural, pure, and non-woven dressing has been made from calcium alginate fibres. It can rapidly absorb and retain wound fluid, to form an integral gellified structure, thereby maintaining an ideal moist wound healing environment. It can also trap and immobilize pathogenic bacteria in the network of gellified fibres Today, with the rapid development of new technologies in bioscience, we expect greater efficacy and further development of dressings or materials that can absorb excess water and reduce the unfavourable effects of occlusion.
Optimal Occlusion Obviously, occlusion alone may damage skin barrier function. Application of chemicals/drugs under occlusion can increase penetration of chemicals and antigens into the skin and therefore can increase dermatitis. Local reactions (i. e. irritation and/or sensitization) of TDDS, typically occlusive patches placed on the skin surface for 1– 7 days to deliver the drugs into the systemic circulation, have been widely reported. However, reactions can be minimized with immunosuppressive agents, antioxidants, local anaesthetics, and other anti-irritant technologies. Advancements in design and construction of protective garments and wound dressings may reduce the level of skin hydration and dermatitis. Application of optimal hydrocolloid patches that absorb water in both liquid and vapor form can also decrease irritation. A natural, pure, and non-woven dressing has been made from calcium alginate fibres. It can rapidly absorb and retain wound fluid, to form an integral gellified structure, thereby maintaining an ideal moist wound healing environment. It can also trap and immobilize pathogenic bacteria in the network of gellified fibres Today, with the rapid development of new technologies in bioscience, we expect greater efficacy and further development of dressings or materials that can absorb excess water and reduce the unfavourable effects of occlusion.

 PENETRATION OF CHEMICAL COMPOUNDS AND DRUGS REF. : EXP DERMATOL 2000: 9: 165– 169 Human skin has many functions and its most apparent is that of a defense organ, both physical and biological. Penetration from outside into the body of any compound is primarily prevented by the corneal layer of the epidermis. This outer layer is just a few micrometers thick, but effectively forms a barrier that is indeed preserving life. Although absorption is not only dependent on penetration, but also on other variables such as skin metabolism, insufficient release from the carrier, partitioning in an unwanted reservoir, without penetration nothing happens. It is important to realize that the human skin has unique properties in this respect and that penetration studies performed in animal models are of limited use for our understanding of the human skin barrier. Essentially, the corneal layer consists of keratin-rich, lipoproteincontaining envelopes and lipid bilayers with hydrophilic regions in between.
PENETRATION OF CHEMICAL COMPOUNDS AND DRUGS REF. : EXP DERMATOL 2000: 9: 165– 169 Human skin has many functions and its most apparent is that of a defense organ, both physical and biological. Penetration from outside into the body of any compound is primarily prevented by the corneal layer of the epidermis. This outer layer is just a few micrometers thick, but effectively forms a barrier that is indeed preserving life. Although absorption is not only dependent on penetration, but also on other variables such as skin metabolism, insufficient release from the carrier, partitioning in an unwanted reservoir, without penetration nothing happens. It is important to realize that the human skin has unique properties in this respect and that penetration studies performed in animal models are of limited use for our understanding of the human skin barrier. Essentially, the corneal layer consists of keratin-rich, lipoproteincontaining envelopes and lipid bilayers with hydrophilic regions in between.
 Most medicaments will pass the epidermal barrier through the intercellular route. As a consequence of its hydrophobic nature, the stratum corneum barrier will allow the penetration of lipid soluble molecules more readily than water-soluble compounds. Strong lipophilic compounds will however be hampered by the hydrophilic regions in the bilayer. Water-soluble molecules may penetrate through an alternative way, the openings of sweat glands and hair follicles. The total surface of these openings amounts to 0. 1% of the total skin surface area, making it probably not significant. The only way to circumvent the properties of the corneal layer is by disrupting it, for example with ultrasound, a method also known as phonopheresis, or with high-voltage electrical pulsing, also known as electroporation. Alternative methods such as stripping the corneal layer using adhesive tape have also been advocated but are not reliable. The use of skin penetration enhancers such as dimethylsulphoxide or carriers such as liposomes have never been confirmed to make a difference.
Most medicaments will pass the epidermal barrier through the intercellular route. As a consequence of its hydrophobic nature, the stratum corneum barrier will allow the penetration of lipid soluble molecules more readily than water-soluble compounds. Strong lipophilic compounds will however be hampered by the hydrophilic regions in the bilayer. Water-soluble molecules may penetrate through an alternative way, the openings of sweat glands and hair follicles. The total surface of these openings amounts to 0. 1% of the total skin surface area, making it probably not significant. The only way to circumvent the properties of the corneal layer is by disrupting it, for example with ultrasound, a method also known as phonopheresis, or with high-voltage electrical pulsing, also known as electroporation. Alternative methods such as stripping the corneal layer using adhesive tape have also been advocated but are not reliable. The use of skin penetration enhancers such as dimethylsulphoxide or carriers such as liposomes have never been confirmed to make a difference.
 In a recent review, it was stated that: ‘‘optimal absorption will occur for molecules that are small, have low melting points. . . ’’. But what is small? An answer to this question is of use for those developing epicutaneous application of compounds to the human skin for destinations varying from topical to systemic treatment to vaccination. The subject of this article is the upper molecular weight (MW) limit for chemical compounds and drugs enabling absorption through the human skin barrier. . We have therefore looked at the MW of common contact allergens and commonly used topical drugs. We propose the 500 Dalton rule, which says that with a MW increasing over 500 Dalton, absorption of molecules through normal human skin rapidly declines.
In a recent review, it was stated that: ‘‘optimal absorption will occur for molecules that are small, have low melting points. . . ’’. But what is small? An answer to this question is of use for those developing epicutaneous application of compounds to the human skin for destinations varying from topical to systemic treatment to vaccination. The subject of this article is the upper molecular weight (MW) limit for chemical compounds and drugs enabling absorption through the human skin barrier. . We have therefore looked at the MW of common contact allergens and commonly used topical drugs. We propose the 500 Dalton rule, which says that with a MW increasing over 500 Dalton, absorption of molecules through normal human skin rapidly declines.
 Dermato-allergology: chemicals causing allergic contact dermatitis are under 712 Dalton The variety of chemicals that are known to lead to allergic contact dermatitis in persons exposed to them forms a true encyclopedia of modern society. Thousands of molecules have been described to be associated with the induction and maintenance of allergic contact dermatitis in a limited or more extensive number of persons. The intrinsic sensitizing capacity of molecules is varying widely. Some compounds rarely lead to clinical contact dermatitis. Others are virtually sensitizing any person whose skin is exposed to it. The routine patch test series, advised by the International Contact Dermatitis Research Group (ICDRG), is used for the diagnosis of contact allergy, and it is composed of the most common sensitizing agents known to mankind. Taking this series of single chemical compounds as well as mixtures of sensitizing agents together, a look at their MW might give us a clue to what size allows penetration through the human skin barrier. A molecule that comes into contact with human skin, but that cannot penetrate the skin barrier in sufficient quantities, will not be a sensitizing agent.
Dermato-allergology: chemicals causing allergic contact dermatitis are under 712 Dalton The variety of chemicals that are known to lead to allergic contact dermatitis in persons exposed to them forms a true encyclopedia of modern society. Thousands of molecules have been described to be associated with the induction and maintenance of allergic contact dermatitis in a limited or more extensive number of persons. The intrinsic sensitizing capacity of molecules is varying widely. Some compounds rarely lead to clinical contact dermatitis. Others are virtually sensitizing any person whose skin is exposed to it. The routine patch test series, advised by the International Contact Dermatitis Research Group (ICDRG), is used for the diagnosis of contact allergy, and it is composed of the most common sensitizing agents known to mankind. Taking this series of single chemical compounds as well as mixtures of sensitizing agents together, a look at their MW might give us a clue to what size allows penetration through the human skin barrier. A molecule that comes into contact with human skin, but that cannot penetrate the skin barrier in sufficient quantities, will not be a sensitizing agent.
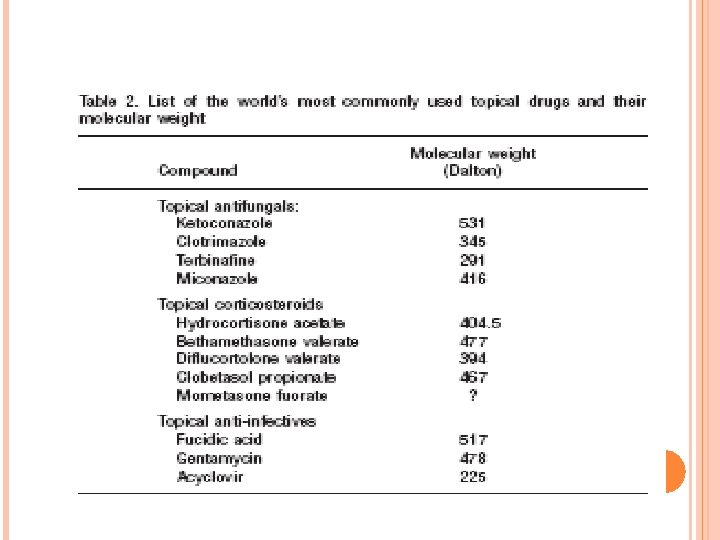
 Molecular weight of the most commonly used topical drugs in dermatotherapy In dermatological therapy, a wide variety of different active molecules is available for topical treatment of individual skin lesions. A list of the most commonly used topical drugs is presented in Table 2. The most commonly used and effective topical drugs in dermatotherapy all have a molecular weight under 500 Dalton, the only exceptions being fusidic acid which is slightly larger with 517 Dalton, and ketoconazole which has a MW of 531 Dalton.
Molecular weight of the most commonly used topical drugs in dermatotherapy In dermatological therapy, a wide variety of different active molecules is available for topical treatment of individual skin lesions. A list of the most commonly used topical drugs is presented in Table 2. The most commonly used and effective topical drugs in dermatotherapy all have a molecular weight under 500 Dalton, the only exceptions being fusidic acid which is slightly larger with 517 Dalton, and ketoconazole which has a MW of 531 Dalton.
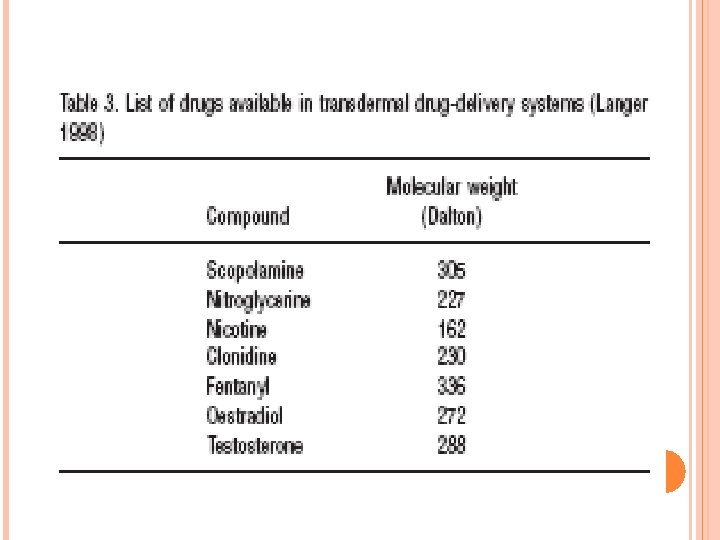
 Molecular weight of drugs used in transdermal drug-delivery systems Certain drugs for systemic use are delivered through the skin for reasons varying from avoiding the liver (destroying drugs when taken orally) to enabling sustained release. Patches with transdermal drug delivery systems are available for at least 7 different drugs. In Table 3, these compounds and their molecular weights are summarized, using the same MW finding strategy as for the topical drugs described earlier. As may be seen from the Table, MWs of drugs used in transdermal drug delivery systems are all well under 500 Dalton, in fact they are all smaller than 350 Dalton.
Molecular weight of drugs used in transdermal drug-delivery systems Certain drugs for systemic use are delivered through the skin for reasons varying from avoiding the liver (destroying drugs when taken orally) to enabling sustained release. Patches with transdermal drug delivery systems are available for at least 7 different drugs. In Table 3, these compounds and their molecular weights are summarized, using the same MW finding strategy as for the topical drugs described earlier. As may be seen from the Table, MWs of drugs used in transdermal drug delivery systems are all well under 500 Dalton, in fact they are all smaller than 350 Dalton.
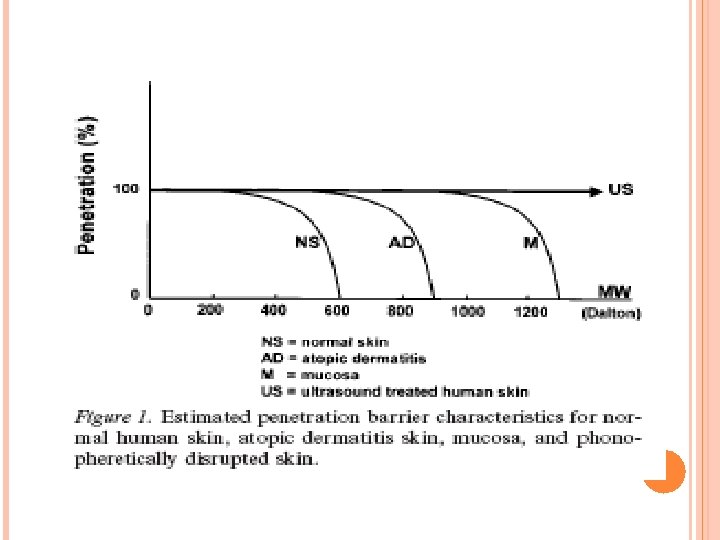
 Concluding remarks The human skin is indeed an effective barrier but it cannot prevent smaller molecules to enter. In Fig. 1, the estimated penetration barrier characteristics for normal human skin, atopic dermatitis skin, mucosa, and phonopheretically disrupted skin are indicated. Somewhere around 500 Dalton is the start of a rapid decline in skin absorption due to molecular size. The barrier is formed by the corneal layer since when absent, such as in mucous membranes, larger molecules may penetrate and thus be effective. Atopic dermatitis forms the exception to the 500 Dalton rule, since it can be managed by topical application of drugs tacrolimus and ascomycin derivatives (822 and 811 Dalton respectively).
Concluding remarks The human skin is indeed an effective barrier but it cannot prevent smaller molecules to enter. In Fig. 1, the estimated penetration barrier characteristics for normal human skin, atopic dermatitis skin, mucosa, and phonopheretically disrupted skin are indicated. Somewhere around 500 Dalton is the start of a rapid decline in skin absorption due to molecular size. The barrier is formed by the corneal layer since when absent, such as in mucous membranes, larger molecules may penetrate and thus be effective. Atopic dermatitis forms the exception to the 500 Dalton rule, since it can be managed by topical application of drugs tacrolimus and ascomycin derivatives (822 and 811 Dalton respectively).
 For pharmaceutical development purposes, it seems logical to restrict the development of new innovative compounds to a MW of under 500 Dalton, when topical dermatological therapy or percutaneous systemic therapy or vaccination is the objective. We therefore propose the 500 Dalton rule for the skin penetration of chemical compounds and drugs. We believe that in drug development, a maximum molecular weight of 500 Dalton should be adhered to, before considering its further development for topical therapy or transcutaneous vaccination in man.
For pharmaceutical development purposes, it seems logical to restrict the development of new innovative compounds to a MW of under 500 Dalton, when topical dermatological therapy or percutaneous systemic therapy or vaccination is the objective. We therefore propose the 500 Dalton rule for the skin penetration of chemical compounds and drugs. We believe that in drug development, a maximum molecular weight of 500 Dalton should be adhered to, before considering its further development for topical therapy or transcutaneous vaccination in man.
 CORRELATION BETWEEN THE TRANSDERMAL PERMEATION OF KETOPROFEN AND ITS SOLUBILITY IN MIXTURES OF A PH 6. 5 PHOSPHATE BUFFER AND VARIOUS SOLVENTS REF. : DRUG DELIVERY, 9: 39– 45, 2002 Introduction “Ketoprofen is a (NSAID) with analgesic and antipyretic actions. it cause, like other NSAIDs, some gastrointestinal and renal side effects. Transdermal drug delivery would be an ideal route for the administration of this drug, to avoid the oral side effects and provide relatively constant drugs levels at the application site for prolonged periods. Transdermal administration of many drugs is often precluded because of the stratum corneum barrier. ”
CORRELATION BETWEEN THE TRANSDERMAL PERMEATION OF KETOPROFEN AND ITS SOLUBILITY IN MIXTURES OF A PH 6. 5 PHOSPHATE BUFFER AND VARIOUS SOLVENTS REF. : DRUG DELIVERY, 9: 39– 45, 2002 Introduction “Ketoprofen is a (NSAID) with analgesic and antipyretic actions. it cause, like other NSAIDs, some gastrointestinal and renal side effects. Transdermal drug delivery would be an ideal route for the administration of this drug, to avoid the oral side effects and provide relatively constant drugs levels at the application site for prolonged periods. Transdermal administration of many drugs is often precluded because of the stratum corneum barrier. ”
 Percutaneous absorption is a passive diffusion process determined essentially by two factors: the dissolution and the diffusion of the drug into the stratum corneum. The contribution of each of these is expressed in Fick’s equation in terms of the partition coefficient of the drug between the stratum corneum and the donor phase (P) and the diffusion constant (D cm 2/h) of the drug in the barrier. P is an index of the affinity of the drug for the vehicle or the skin and can be optimized by decreasing its solubility in the vehicle. D is related to the characteristics of the drug. It could change using Enhancers that fluidize the lipids or induce structural changes in Proteins of the stratum corneum or using cosolvents that increase the partition tendencies of the permeant in the rate-limiting lipid phase.
Percutaneous absorption is a passive diffusion process determined essentially by two factors: the dissolution and the diffusion of the drug into the stratum corneum. The contribution of each of these is expressed in Fick’s equation in terms of the partition coefficient of the drug between the stratum corneum and the donor phase (P) and the diffusion constant (D cm 2/h) of the drug in the barrier. P is an index of the affinity of the drug for the vehicle or the skin and can be optimized by decreasing its solubility in the vehicle. D is related to the characteristics of the drug. It could change using Enhancers that fluidize the lipids or induce structural changes in Proteins of the stratum corneum or using cosolvents that increase the partition tendencies of the permeant in the rate-limiting lipid phase.
 Aim of the work “To find any correlation between the permeability of ketoprofen through the skin and its solubility in mixtures of water and co-solvents, To choose the co-solvent that best enhances the permeability of ketoprofen in the development of hydrophilic topicals. ”
Aim of the work “To find any correlation between the permeability of ketoprofen through the skin and its solubility in mixtures of water and co-solvents, To choose the co-solvent that best enhances the permeability of ketoprofen in the development of hydrophilic topicals. ”
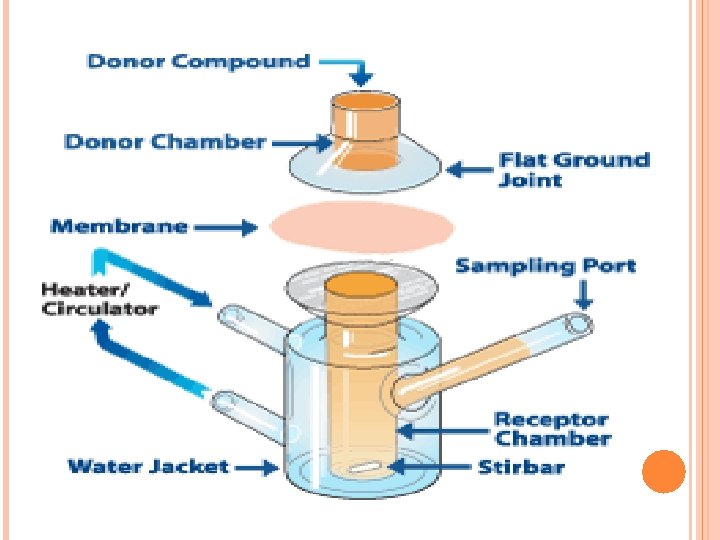
 Materials and Methods Drug: Ketoprofen, Solvents: ethoxyethylendiglycol, ethyl alcohol, Propylen glycol, absolute, isopropyl alcohol, glycerol 90% and PEG 400, Diffusion barrier: Porcine skin, the skin was surgically removed from the ears of freshly killed male pigs, Diffusion instrument: standard Franz diffusion cells having 0. 64 cm 2 diffusion area, and 4. 8 ml receptor compartment volume. (See figure) Analytical method: HPLC, drug detection at 276 nm.
Materials and Methods Drug: Ketoprofen, Solvents: ethoxyethylendiglycol, ethyl alcohol, Propylen glycol, absolute, isopropyl alcohol, glycerol 90% and PEG 400, Diffusion barrier: Porcine skin, the skin was surgically removed from the ears of freshly killed male pigs, Diffusion instrument: standard Franz diffusion cells having 0. 64 cm 2 diffusion area, and 4. 8 ml receptor compartment volume. (See figure) Analytical method: HPLC, drug detection at 276 nm.
 Solubility Studies at 370 C Ketoprofen + p. H 6. 5 phosphate buffer 0. 5 M + different co-solvents at different concentrations. Excess drug >> heat to 500 C >> equilibrated at 370 C for 24 h. Aliquots of the saturated systems were filtered, diluted with mobile phase, and analyzed by HPLC.
Solubility Studies at 370 C Ketoprofen + p. H 6. 5 phosphate buffer 0. 5 M + different co-solvents at different concentrations. Excess drug >> heat to 500 C >> equilibrated at 370 C for 24 h. Aliquots of the saturated systems were filtered, diluted with mobile phase, and analyzed by HPLC.
 In vitro Diffusion Study at 370 C Diffusion cell was filled with fresh PBS, The solution in the receptor compartments was continuously stirred at 600 rpm using a Teflon coated magnetic stirrer. The porcine skin, 1± 0. 1 mm thick, was clamped between the donor and receiving compartments. 1 ml of the saturated solutions or the gel was placed in the donor compartment. The amount of ketoprofen diffused through porcine skin was determined by removing aliquots of 2 ml from the receptor compartments using a syringe and immediately replacing with the same volume of PBS (kept at 370 C). The sampling schedule was 0. 5, 1, 2, 4, 6, and 8 h.
In vitro Diffusion Study at 370 C Diffusion cell was filled with fresh PBS, The solution in the receptor compartments was continuously stirred at 600 rpm using a Teflon coated magnetic stirrer. The porcine skin, 1± 0. 1 mm thick, was clamped between the donor and receiving compartments. 1 ml of the saturated solutions or the gel was placed in the donor compartment. The amount of ketoprofen diffused through porcine skin was determined by removing aliquots of 2 ml from the receptor compartments using a syringe and immediately replacing with the same volume of PBS (kept at 370 C). The sampling schedule was 0. 5, 1, 2, 4, 6, and 8 h.
 RESULTS IN ALL EXPERIMENTS, THE PERMEATION PROFILES SHOWED A CURVED PORTION, CORRESPONDING TO A LAG PHASE, FOLLOWED BY A LINEAR PORTION, WHICH MEANS THAT AN APPARENT STEADY STATE OF KETOPROFEN PENETRATION WAS ATTAINED.
RESULTS IN ALL EXPERIMENTS, THE PERMEATION PROFILES SHOWED A CURVED PORTION, CORRESPONDING TO A LAG PHASE, FOLLOWED BY A LINEAR PORTION, WHICH MEANS THAT AN APPARENT STEADY STATE OF KETOPROFEN PENETRATION WAS ATTAINED.
 Data Analysis The in vitro skin flow was determined from Fick’s law of diffusion: Js = (1/A)(d. M/dt) = Kp ∆C eq. 1 Kp = (D* P)/h eq. 2 Lag time = h 2/6 D eq. 3 In all the experiments, the concentration in the donor cell remained constant, the receiver cell concentrations did not exceed 10% of the donor cell concentrations, and ∆C was assumed to be equal to the donor cell concentrations. The steady-state flux Js was determined from the slope of the linear portion of the cumulative amount permeated per unit area versus time plot. Js& the permability constant, Kp determined from eq. 1. The lag time was determined by extrapolating the linear portion of the curve to the abscissa. D was approximated indirectly from the lag time (eq. 3), P is then determined using eq. 2
Data Analysis The in vitro skin flow was determined from Fick’s law of diffusion: Js = (1/A)(d. M/dt) = Kp ∆C eq. 1 Kp = (D* P)/h eq. 2 Lag time = h 2/6 D eq. 3 In all the experiments, the concentration in the donor cell remained constant, the receiver cell concentrations did not exceed 10% of the donor cell concentrations, and ∆C was assumed to be equal to the donor cell concentrations. The steady-state flux Js was determined from the slope of the linear portion of the cumulative amount permeated per unit area versus time plot. Js& the permability constant, Kp determined from eq. 1. The lag time was determined by extrapolating the linear portion of the curve to the abscissa. D was approximated indirectly from the lag time (eq. 3), P is then determined using eq. 2
 Relation between solubility and D: When statistical analysis was carried the solubilities of the ketoprofen in the different mixtures and D values, statistical significance (p < 0. 05) was not recognized. This means that the presence of the cosolvents does not change the diffusion rate of ketoprofen. >>> no realtion.
Relation between solubility and D: When statistical analysis was carried the solubilities of the ketoprofen in the different mixtures and D values, statistical significance (p < 0. 05) was not recognized. This means that the presence of the cosolvents does not change the diffusion rate of ketoprofen. >>> no realtion.
 RELATION BETWEEN PERMEABILITY CONSTANT KP AND KETOPROFEN SOLUBILITIES IN THE SATURATED SOLUTIONS>>> INVERSE PROPRTIONALITY .
RELATION BETWEEN PERMEABILITY CONSTANT KP AND KETOPROFEN SOLUBILITIES IN THE SATURATED SOLUTIONS>>> INVERSE PROPRTIONALITY .
 KETOPROFEN SOLUBILITIES IN THE SATURATED SOLUTIONS>>> INVERSE PROPRTIONALITY .
KETOPROFEN SOLUBILITIES IN THE SATURATED SOLUTIONS>>> INVERSE PROPRTIONALITY .
 Using saturated solutions Highest Kp value& highest flux value Pb+10% glycerol vs PB+40% Ethanol Solubility 7434 vs 71151 ug/ml Kp 7. 4 vs 1. 4 cm/h Flux, J 55 vs 98. 6 ug/cm 2 h D/1000 34 vs 34. 8 cm 2 h Lag time 1. 73 vs 1. 72 h i. e the solvent can modify both Kp and J.
Using saturated solutions Highest Kp value& highest flux value Pb+10% glycerol vs PB+40% Ethanol Solubility 7434 vs 71151 ug/ml Kp 7. 4 vs 1. 4 cm/h Flux, J 55 vs 98. 6 ug/cm 2 h D/1000 34 vs 34. 8 cm 2 h Lag time 1. 73 vs 1. 72 h i. e the solvent can modify both Kp and J.
 Conclusion Permeation of ketoprofen can be modulated using various co-solvents. Since D values of the in vitro permeation studies of the saturated solutions are not significantly altered in the tested mixtures, the variation in permeability coefficient of ketoprofen across skin is a result of a variation in partitioning of the drug from the vehicle into the stratum corneum. The present investigation demonstrates that the permeation ability of ketoprofen across skin can be successfully predicted from the knowledge of only the physiochemical parameter of solubility into an aqueous mixture of phosphate buffer and a co-solvent. This is possible because the permeability constant for ketoprofen is inversely proportional to the solubility in the vehicle.
Conclusion Permeation of ketoprofen can be modulated using various co-solvents. Since D values of the in vitro permeation studies of the saturated solutions are not significantly altered in the tested mixtures, the variation in permeability coefficient of ketoprofen across skin is a result of a variation in partitioning of the drug from the vehicle into the stratum corneum. The present investigation demonstrates that the permeation ability of ketoprofen across skin can be successfully predicted from the knowledge of only the physiochemical parameter of solubility into an aqueous mixture of phosphate buffer and a co-solvent. This is possible because the permeability constant for ketoprofen is inversely proportional to the solubility in the vehicle.