Хаценко Л.А. Хромосоми.ppt
- Количество слайдов: 53




1. Спадкові хвороби, їх визначення і принципи класифікації. 2. Генні хвороби. 3. Хромосомні хвороби: а) гетероплоїдія за аутосомами: хвороби Дауна, Едвардса, Патау; б) гетероплоїдія за гетеросомами: синдром Шерешевського-Тернера, Клайнфельтера, полісомія - х, полісомія - у. 4. Мультифакторіальні хвороби.

Спадкові захворювання виникають внаслідок пошкодження структури і функції генетичного апарату клітини. Спадковими називаються захворювання, зумовлені зміною генів або хромосом у гаметах батьків, в результаті чого зигота з початку виникнення несе генну , хромосомну або геномну мутацію. Слід розрізняти спадкові захворювання від уроджених, які з'являються ще в процесі ембріогенезу. Спадкові хвороби становлять близько 75% уроджених. Серед загальної захворюваності населення спадкова становить 15 - 25%. Тепер відомо близько 3500 спадкових захворювань і кількість їх зростає.
, які викликаються променевою, тепловою енергією,")
Спадкові захворювання виникають внаслідок змін спадкового апарату клітин (мутацій), які викликаються променевою, тепловою енергією, хімічними речовинами (у тому числі й лікарськими) і біологічними факторами (вірусами, глистяними токсинами тощо ). Кожна людина має 5 -10 потенціально шкідливих генів, які передаються нащадкам разом з нормальними генами.

Кожна фенотипова здорова людина носить приблизно 8 -10 мутагенних генів. Всі відомі захворювання поділяються: 1. Набуті (опіки, травми, інфекційні хвороби); 2. Спадкові (патологічна спадковість) гемофілія, синдром Дауна та інші. 3. Мультифакторіальні (спадкова схильність) цукровий діабет, виразкова хвороба шлунка та інші.
 – 3 %.")
Усі спадкові хвороби можна поділити на три групи: 1. Генні (моногенні) – 3 %. 2. Хромосомні хвороби (синдром) – 1%. 3. Полігенні або мультифакторіальні.

Причиною молекулярних, так само як і хромосомних захворювань є пошкодження нитки ДНК у статевих або в соматичних клітинах. Різниця між цими захворюваннями в тому, що при молекулярних захворюваннях в молекулі ДНК пошкоджений один або кілька генів, тобто порівняно невелика частина ДНК, а тому великих морфологічних змін у хромосомах не відбувається. Генетичні зміни мають молекулярний характер, а тому і називається молекулярними спадковими захворюваннями. При хромосомних захворюваннях зміни в молекулі ДНК займають більші ділянки, через це формування хромосом проходить неправильно. В цьому випадку під мікроскопом помітні якісні (морфологічні) або кількісні зміни хромосом.

Класифікація моногенних хвороб: 1. Генетична класифікація враховує АД-, АР-, ХД-, ХР-, У-зчеплені хвороби. 2. Клінічна класифікація враховує, який орган або система ушкоджені. 3. Патогенетична класифікація, враховує патогенетичну ланку.

Більшість спадкових моногенних захворювань – це дефект обміну речовин. За класифікацією ВООЗ вони поділяються на 11 груп. Їх відомо більш як 1000. Найбільшу групу становлять захворювання, спричинені порушенням обміну речовин, які представлені чотирма класами білків: • білками - ферментами; • структурними; • транспортними; • циркулюючими. Більшість спадкових дефектів обміну - ферментопатії, тобто порушення будови білків – ферментів, які беруть участь в обміні тих чи інших речовин. При цьому в організмі спостерігається дефіцит кінцевого продукту обміну і накопичення проміжних продуктів.

Фенілконурія - це хвороба, яка спричинена порушенням амінокислотного обміну. В організмі людини а-к фенілаланін під впливом ферменту фенілаланін- гідроксілази перетворюється в тирозин, а останній через ряд проміжних продуктів обміну - в тироксин, меланін, адреналін. При відсутності ферменту він накопичується в тканинах і перетворюється в інші продукти (фенілпіровиноградну кислоту та ін. ). Вони є токсичними продуктами для нервової системи, особливо до головного мозку в період його формування. Внаслідок цього у дитини спостерігається розумова відсталість в вигляді імбецільності та ідіотії. Нестача тирозину призводить до зменшеного утворення адреналіну й меланіну. Тому в цих дітей відзначається зменшення пігментації волосся, райдужки і схильність до артеріальної гіпотензії.

Вперше фенілкетонурію описав норвежський лікар Ф. Фелінг у 1934 р. , який за допомогою Fе. СL 3 виявив у сечі двох розумово відсталих дітей фенілпіровиноградну кислоту. Діти народжуються нормальними, добре набирають масу, але вже на другому півріччі життя у них виявляються відставання у психічному розвитку. Діти стають сонливими, в'ялими, не фіксують погляд на речах, не прагнуть до спілкування з батьками, у них послаблена увага. З'являється блювота, різні форми дерматиту. Більшість цих дітей світловолосі, з голубими очима. Далі виявляється відставання і в фізичному розвитку. Затримується прорізування зубів, розвиток мови тощо.

У материнському молоці фенілаланіну менше, ніж у коров'ячому, тому чим довше дитина перебуває на грудному вигодовуванні, тим пізніше проявляється захворювання. Розроблені ефективні дієти для харчування дітей, хворих на фенілкетонурію. Вони базуються на заміні натуральних білкових продуктів молока, м'яса, яєць гідролізатами, які мають мінімальну кількість фенілаланіну при нормальній концентрації інших амінокислот. Цю дієту треба назначати до 5 -річного віку. Однак лікування треба проводити під наглядом лікаря, контролюючи конц. фенілаланіну в крові. Оптимальне вважається його конц. 0, 03 - 0, 08 г/л (у здорових людей - 0, 01 - 0, 04 г/л).

Повне припинення надходження фенілаланіну несприятливо впливає на ріст і розвиток дитини, тому що фенілаланін - незамінна амінокислота. Додатково таким дітям призначають вітаміни, особливо вітамін С Тепер виготовляється декілька препаратів: лофенолак, мінафен, кетоніл, цимогран, гіпофенат. Лікування зазначеними препаратами тим ефективніш, чим раніше воно розпочато, поки ще не почалося руйнування головного мозку.
 Пробірку з сечею добавляють 6 -10 крапель 10%")
Діагностика фенілкетонурії. 1. Реактив Фелінга. а) Пробірку з сечею добавляють 6 -10 крапель 10% розчину Fе. СL 3. За наявності в сечі фенілаланіну вона забарвлюється синьо-зелений колір. Через 5 - 10 хв. зелений колір поступово світлішає, а тому реакцію треба враховувати відразу після проведення досліду. б) Пелюшкова проба. в) Проба з індикаторним папірцем. Реакція Фелінга стає позитивною тоді, коли в крові концентрація фенілаланіну не менша за 01 - 15 г/л. Це звичайно буває у віці 2 -5 тижнів життя дитини.
. До")
2. Більш специфічною є реакція з 2, 4 – динітрофенілгідразином (2, 4 -ДНФГ). До 1 - 2 мл. сечі добавляють 0, 3% розчину 2, 4 ДНФГ. Через 1 -3 хв. в пробірці з'являється каламуть яскраво-жовтого кольору, яка не зникає протягом доби. З. Визначення концентрації фенілаланіну в крові методом хроматографії на папері або іншими методами. 4. Амінокислотні аналізатори.

5. Мікробіологічний тест Гатрі. Диски фільтрувального паперу змочують кров'ю дитини. Кров беруть з пуповини дитини після народження або одержують уколом у п'ятку. Ці диски автоклавують для зруйнування еритроцитів і коагуляції білків. У чашки Петрі висівають культуру Вас Subtilis, в яку добавляють інгібітор росту-аналог фенілаланіну і диски, які змочені кров'ю дитини. Якщо в крові є фенілаланін, він нейтралізує аналог фенілаланіну і навколо цього диска спостерігається ріст культури мікробів. Навколо дисків, де немає фенілаланіну, росту мікробів немає. Цим методом можна дуже рано виявити захворювання.

6. Мікробіологічний метод визначення амінокислот за допомогою ауксотрофних штамів кишкової палочки. Цей метод застосовують під час масового обстеження дітей. Кров гаданого хворого можна в змочених і висушених дисках направляти поштою у великі біохімічні лабораторії. Для того, щоб дати позитивну доповідь відносно фенілкетонурії на основі цього методу, треба, щоб конц. фенілаланіну в крові досягла рівня 0, 15 г/л і вище, а в сечі – 1 г/л.

7. Гетерозиготних носіїв можна виявити навантажувальним методом: введення фенілаланіну з розрахунку ОД г на 1 кг маси тіла і наступним визначенням фенілаланіну в крові. У здорових людей конц. фенілаланіну зростає за першу годину в 4 - 5 разів, а у гетерозиготних носіїв фенілкетонурії – у 10 і більше разів. Частота народження хворих на фенілкетонурію 1: 20000 пологів. Лікування:

До спадкових захворювань порушення вуглеводного обміну належить: глікогеноз, галактоземія та інші захворювання. Галактоземія. Галактоза - це складова частина молочного цукру (лактози). В організмі вона за допомогою ферменту галактозо - 1 фосфат - уридилтрансферази перетворюється в галактозо - 1 фосфат, який далі перетворюється в глюкозо-6 фосфат, що входить до метаболічного циклу глюкози. При галактоземії у дитини відзначається накопичення в організмі галактозу токсично впливає на тканини. У дитини розвивається цироз печінки, уражуються нирки, розвивається рання катаракта; диспептичні розлади, діти худнуть, з'являється жовтяниця, затримується психічний розвиток. Ці діти вмирають у перші місяці життя, якщо не буде своєчасно призначене необхідне лікування. Якщо дитина залишається живою, то у неї відзначається мікроцефалія, знижується тонус м'язів, з'являються судоми, гепато - і спленомегалія, розвивається анемія.

Рання діагностика захворювання проводиться на ауксотрофних мікробах, а також хроматографічним визначеним конц. амінокислот у крові дитини. Захворювання успадковується А-р. Лікування: відлучення дитини від груді і переведення на годування коров'ячим молоком ( в якому галактози менше ніж у жіночому), а також на спеціальну дієту.

Синдром Гурлера При порушенні синтезу мукополісахаридів вони відкладаються в клітинах організму і розвивається мукополісахаридоз. Вперше було описано Гурлером у 1919 році і було визначене як гаргоілізм, тому що голова хворих нагадує гаргоїлів потворних фігур на паризькому соборі. Захворювання діагнозується на другому році життя дитини шляхом виявлення в сечі підвищеної концентрації кислих мукополісахаридів, рентген: виявляють множинні дефекти скелету.

Воно виявляється розумовою відсталістю, скелетними змінами: кіфозом, випинанням лоба, плоским носом, гіпертелоризмом, потовщенням губ, дрібними зубами, збільшеним язиком, деформованими вушними раковинами, густим жорстким волоссям, коротким тулубом, деформованою грудною кліткою, збільшенням поперекового і грудного кіфозу, потовщеними і розширеними епіфізами довгих кісток. Пальці рук при синдромі Гурлера набувають напівзігнутого положення, відзначається деяка скутість хворих, часті пупкові грижі, гепатоспленомегалія, серцеві вади, помутніння рогівки, нерідко - вроджена глаукома. Прогноз несприятливий. Лікування неефективне.

До спадкових порушень обміну ліпідів належить хвороби Тея-Сакса, Гоше, Німана-Піка. Це лізосомні захворювання. Через дефекти лізосомних ферментів, які беруть участь у метаболізмі ліпідів, останні накопичуються всередині клітин і викликають їхню загибель. Ліпіди - це складні сполуки ненасиченого аміноспирту, жирних кислот і вуглеводів. Перетворення одного ліпіду в інший здійснюється за допомогою відповідного ферменту. Дефект того чи іншого ферменту призводить до накопичення всередині клітин відповідного ліпіду, що називається ліпідозом.

Хвороба Гоше. В клітинах головного мозку і внутрішніх органах накопичується глюкоцереброзид (сполука цераміду з глюкозою). Виділяють гостру, підгостру і хронічну форми захворювання. При гострих формах спостерігається м'язова ригідність, порушення зору, психічна деградація, остеопороз кісток, стоншення картикального шару їх, деформація стегон за типом "пляшок". Часті переломи кісток через розростання клітин Гоше в кістковому мозку.

Часті тромбопенії, носові кровотечі. Збільшений живіт. Такі діти звичайно вмирають на першому році життя від виснаження. При підгострій формі захворювання всі симптоми мають більш м'який характер. При хронічній (вісцеральній) формі пошкоджуються внутрішні органи без втягнення в процес головного мозку. Хвороба Гоше - гетерогенне захворювання. Описані як А-Р, так і А -Д форми.

Хромосомні хвороби, що пов'язані зі зміною числа аутосом. Хвороба Дауна. Вперше описана у 1866 р. англійським лікарем Л Дауном. Французським вченим І. Лежен у 1959 р. виявив у каріотипі хворих зайву 21 -у хромосому. Частота народження дітей, хворих на хворобу Дауна, становить 1, 15 -5/1000. Причини остаточно не з'ясовані. 1. Перенесені матір'ю перед заплідненням інфекційні захворювання (гепатит, токсоплазмоз тощо). 2. Із збільшенням віку матері ризик народження дитини з хворобою Дауна збільшується. 19 років 1 : 1640 40 -41 1 : 84 після 45 1: 31 (Лазюк, 1979)

Симптоми: кругла голова зі сплющеною потилицею, покатим вузьким лобом, плоским обличчям, монголоїдним розрізом очних щілин, епікантом, товстими губами, широким сплющеним язиком з глибокою поздовжньою борозною. Вушні раковини зменшені, з прирослою мочкою. М'язова гіпотонія, жаб'ячий живіт (у маленьких дітей), розхитаність суглобів, куряча або лійкоподібна грудна клітина. Кінцівки: акромікрія (укорочення і розширення кисті і ступні); викривлення мізинця ( линодактілія), на долоні одна поперечна борозна. На райдужній оболонці є світлі плями, рано розвивається катаракта.

Неправильний ріст зубів, високе піднебіння, рум'янець на щоках. Волосся на голові м'яке, пряме. Порушені внутрішні органи особливо серце, шлунковокишковий канал та ін. У хворих спостерігається глибока розумова відсталість. Вони слухняні, добре піддаються навіюванню. Писати і читати вони ще можуть навчитися, але лічити не вміють за недорозвиттям мозку. Є випадки вагітності і родів у жінок, які хворіли на хворобу Дауна (47, 21 +). Приблизно у ½ їхніх дітей також розвивалася хвороба Дауна. Батьківство у чоловіків, які хворіють на хворобу Дауна, не описано. Напевне трисомія 21 -ї хромосоми у чоловіків призводить до безплідності.

Синдром Патау, трисомію за 13 -хром. 47, 13+ Описана Патау у 1960 році. Симптоми: мікроцефалія, низький спадистий лоб, вузькі очні щілини, вузьке розташування вушних раковин, розщеплені верхня губа і піднебіння, полідактилія, дефекти ссс і внутрішніх органів, недорозвинуті передні відділи мозку. Такі діти вмирають у перші три місяці життя або протягом року.

Синдром Едвардса, трисомія за 18 хром. 47, 18+ Частіше цей синдром спостерігається у хлопчиків. Дефект черепа і скелета. Діти народжуються переношеними. В асфіксії, з доліхоцефалією, вузьким лобом, виступаючою потилицею, розщепленим піднебінням, недорозвинутою нижньою щелепою, деформованими і низько розміщеними вушними раковинами. Стопа має форму качалки, пальці ніг укорочені. Шкіра дуже рухлива, через що утворюються зморшки на шиї та інших частинах тіла. Очі маленькі. Значні дефекти внутрішніх органів (серця, шлунково-кишкового каналу), у хлопчиків відмічається крипторхізм, а у дівчаток – гіпертрофія клітора. Хлопчики звичайно помирають незабаром після народження, а дівчатка живуть до року. Моносомія за будь-якою аутосомою з життям не сумісна, а тому живонароджені плоди невідомі.

Хромосомні хвороби, що пов'язані зі зміною статевих хромосом. Сумісними з життям є зміни кількості статевих хромосом - моносомія і трисомія. Це можна пояснити тим, що в статевих хромосомах активність генів виявляється переважно в процесі ембріогенезу, а потім вона знижується. В організмі дорослих активних генів, що містяться в статевих хромосомах, мало.

Синдром Шерешевського-Тернера. Вперше описано у 1925 році Шерешевським, в 1938 р. – Тернером. У 1959 році Форд встановив, що у цих хворих є тільки одна статева х -хромосома. Каріотип 45, хо. Ці жінки низького зросту, з статевим інфантилізмом. У них аномальні статеві органи, вузька піхва, недорозвинуті матка і яєчник, гіпертрофований клітор, мізерне оволосіння на лобку. У них не буває менструацій, або вони одноразові. Грудні залози відсутні. На місце їх інколи є складки жиру. Соски недорозвинуті, широко розставлені і непігментовані. Вушні раковини деформовані, вузько розміщені, шия коротка, з її боків видно широку шкірну зморшку, яка йде від соскоподібних відростків до надпліччя. Волосся на потилиці росте низько. Часто відмічається епікант, вузьке і високе тверде піднебіння.

Спостерігаються зміни кінцівок: широкі долоні, короткий безіменний палець, короткий викривлений мізинець, деформовані глибоко посаджені нігті, на кінчиках пальців переважають кругові візерунки. Може спостерігатись стеноз легеневого стовбура, незарощення міжшлункової перегородки серця, підковоподібна нирка. Інтелект у хворих порушений мало, або зовсім не порушений. У хворих різко знижене виділення естрогенів і підвищене виділення гонадотропіну. Причина народження дітей з синдромом Шерешевського-Тернера не з'ясована. Вік батьків при цьому значення не має. Винними можуть бути як батько, так і мати.

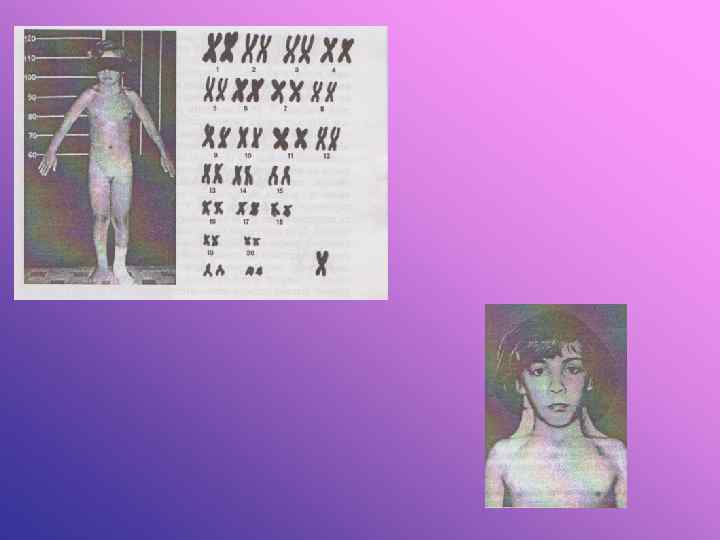
Синдром полісомії за Х-хромосомою у жінок. Каріотип 47, ххх. Змін за фенотипом може й не бути, тому що дві х-хромосоми спіралізовані і представлені статевим х-хроматином. Може відзначатись розумова відсталість. Можуть мати здорових нащадків, через те що 1/2 їхніх гамет має нормальний набір хромосом. Описані випадки з 4 і 5 - хромосомами. Чим більше Х-хромосом в каріотипі, тим більше виражений дефект розумового розвитку, а також зміни фенотипу і статевий інфантилізм. Такі жінки високого зросту із значними змінами скелета, викривленням хребта, депігментованими плямами, гіпоплазія матки, безплідність, часто буває передчасний клімакс, схильність до шизофренії. Буває порушення менструального циклу.

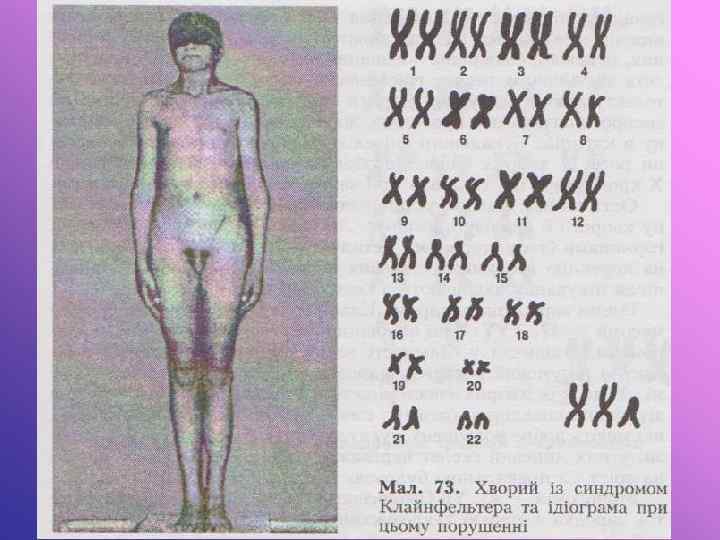
Синдром Клайнфельтера. Вперше описаний Клайнфельтером у 1924 р. У 1956 р. Брігс, Барр виявили у них зайву х-хромосому. Каріотип 47, хху. В клітинах букального епітелію виявлений статевий х-хроматин. Діагноз до статевої зрілості може бути не виявленим. При статевій зрілості відзначаються ознаки євнухоїдності. Недорозвинуті статеві залози при нормальному розвитку статевого члена, відсутня волосатість на обличчі, гінекомастія, відкладання жиру на стегнах за жіночим типом. Вони звичайно високого зросту за рахунок подовження ніг, тулуб відносно короткий. Волосся на лобку росте за жіночим типом, у хворих високий голос. Під час гістологічного дослідження яєчок відзначається підвищене виділення фолікуліну. Сперматогенез відсутній, тому вони неплідні. Дебільність різного ступеня. Чим більше х-хромосом в каріотипі, тим більше виражений ступінь дебільності та інші симптоми синдрому.
")
Мікроцефалія (недорозвинення головного мозку та черепа)
")
Зовнішній вигляд дитини з хворобою Німанна – Піка (смертельна)
 Хвороба Тея - Сакса")
Хвора на мукополісахаридоз (синдром Санфіліппо) Хвороба Тея - Сакса
 Пікнодизостоз (дистальні фаланги пальців кистей укорочені)")
Хворий із синдромом розумової відсталості (розрив Х-хромосоми) Пікнодизостоз (дистальні фаланги пальців кистей укорочені)

Хвора на муковісцидоз


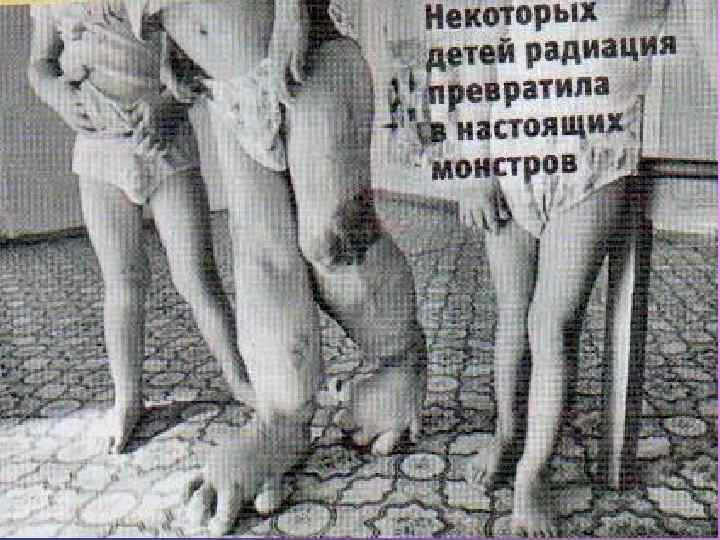
Вся правда про мутантів Чорнобиля







Хаценко Л.А. Хромосоми.ppt