
Хромосомные болезни План занятия 1. Геномные и хромосомные


Хромосомные болезни

План занятия 1. Геномные и хромосомные заболевания. 2. Синдромы их диагностика и исследование. 3. Мозаики. 4. Численные хромосомные аномалии. 5. Структурные хромосомные аномалии.

Наследственное заболевание — заболевание, возникновение и развитие которого связано с дефектами в наследственном аппарате клеток, передаваемыми по наследству через гаметы.
 ГЕНОМНЫЕ (кратное изменение хромосомного набора – полиплоидия)")
ГЕННЫЕ (изменения на уровне отдельных нуклеотидов) ГЕНОМНЫЕ (кратное изменение хромосомного набора – полиплоидия) ХРОМОСОМНЫЕ (перемещение участков хромосом или их обмен) СОМАТИЧЕСКИЕ (не передаются по наследству) ТИПЫ МУТАЦИЙ

Мутагены — вещества, вызывающие изменения ДНК, генов МУТАГЕНЫ РЕНТГЕНОВСКИЕ ЛУЧИ ЯДОВИТЫЕ ВЕЩЕСТВА (КОЛХИЦИН) КАНЦЕРОГЕННЫЕ ВЕЩЕСТВА НЕКОТОРЫЕ ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ

Типы наследственности 1. Аутосомно-доминантный тип наследования: а. При достаточном числе потомков признак обнаруживается в каждом поколении б. Редкий признак наследуется примерно половиной детей в. Потомки мужского и женского пола наследуют этот признак одинаково г. Оба родителя в равной мере передают этот признак детям 2. Аутосомно-рецессивный тип наследования: а. Признак может передаваться через поколение даже при достаточном числе потомков б. Признак может проявиться у детей в отсутствие его у родителей. Обнаруживается тогда в 25% случаев у детей в. Признак наследуется всеми детьми, если оба родителя больны г. Признак в 50% развивается у детей, если один из родителей болен д. Потомки мужского и женского пола наследуют этот признак одинаково 3. Наследование, сцепленное с Х хромосомой, если ген, контролирующий проявления признака, - рецессивный: а. Мужчины наследуют чаще, чем женщины б. Наследуют такой признак девочки только от отца в. В браках, где оба супруга здоровы, могут родиться дети, имеющие его, при этом он наследуется 50% сыновей и 100% здоровых дочерей г. Прослеживается чередование больных мужчин в поколениях: где их больше, где - меньше 4. Наследование, сцепленное с Х хромосомой, если ген, контролирующий проявления признака, - доминантный: а. Мужчины наследуют реже, чем женщины б. Если признак только у супруги, то наследуют его все дети (мать гомозиготная), или половина детей (мать гетерозиготная) в. Если только у супруга, то наследуют все лица женского пола 5. Наследование, сцепленное с Y хромосомой: а. Страдают только сыновья, в каждом поколении проявляется, если отец болен.

Хромосомные болезни – наследственные заболевания, обусловленные изменением числа или структуры хромосом. Частота хромосомных болезней среди новорождённых детей около 1%. Многие изменения хромосом несовместимы с жизнью и являются частой причиной спонтанных абортов и мертворождений. При спонтанных абортах обнаружено около 20% эмбрионов с аномальными кариотипами (хромосомными наборами).

Хромосомные наследственные болезни Все хромосомные болезни с нарушением состояния хромосом можно разделить на две большие группы: изменение числа хромосом при сохранении структуры последних (геномные мутации); изменением структуры хромосомы (хромосомные мутации). Численные нарушения могут состоять в изменении плоидности хромосомного набора и в отклонении числа хромосом от диплоидного по каждой их паре в сторону уменьшения (моносомия) или увеличения (полисомия).

Причины болезней Наследственные болезни связанные с нарушением плоидности вызванные нарушением числа хромосом связанные с изменением структуры хромосом. ХРОМОСОМНЫЕ БОЛЕЗНИ
 отдельных")
Основные типы аномалий: численные изменения хромосомного набора; структурные изменения (аберрации) отдельных хромосом.

Изменение числа хромосом происходит в результате нерасхождения их в мейозе или при делении клеток на ранней стадии развития оплодотворённого яйца. Нерасхождению хромосом при первых делениях оплодотворённого яйца способствует, например, высокий возраст матери. Хромосомные аберрации обусловливаются физическими (ионизирующее излучение) и химическими (например, лекарственные препараты с мутагенным эффектом) факторами; вирусами (краснухи, вирусного гепатита, ветряной оспы и др.), антителами и различными расстройствами метаболизма. Хромосомные болезни могут быть связаны с излишком генетического материала (полисемия - наличие одной или нескольких добавочных хромосом; полиплоидия; дупликация); с утратой части генетического материала (нуллисомия, моносомия, делеция); с хромосомными перестройками (транслокация; различные перестановки участков хромосом). Различают также группы Х. б., обусловленных изменениями половых и неполовых хромосом. Изменение числа хромосом происходит в результате нерасхождение их в мейозе или при делении клеток на ранней стадии развития оплодотворённого яйца. Возраст мамы будущего ребенка. Ионизирующее излучение. Лекарственные препараты с мутагенным эффектом, алкоголь. Вирусы (краснухи, вирусного гепатита, ветряной оспы и др.). Антителами и различными расстройствами метаболизма.

Геномные болезни могут быть связаны с излишком генетического материала (полисомия - наличие одной или нескольких добавочных хромосом; полиплоидия; дупликация); с утратой части генетического материала (нулисомия, моносомия, делеция); с хромосомными перестройками (транслокация; различные перестановки участков хромосом). Различают также хромосомные болезни, обусловленные изменениями половых и неполовых хромосом.

Хромосомные заболевания сопровождаются: Многочисленными поражениями скелета Нарушением психики Врожденными пороками внешних и внутренних органов Задержкой роста Поражением нервной, эндокринной и др. систем Сниженной регенераторной функцией Повышенной заболеваемостью и смертностью

ДИАГНОСТИЧЕСКИЕ ПРИЗНАКИ ХРОМОСОМНЫХ АНОМАЛИЙ: А — комплекс признаков, что позволяют заподозрить хромосомную аномалию: физическое недоразвитие, ряд дисморфий мозгового и лицевого черепа, плоскостопие, ряд аномалий внутренних органов (сердца, лёгких, почек) В - признаки, которые встречаются в основном при определённых хромосомных заболеваниях. Их комбинация позволяет в большинстве случаев диагностировать хромосомную аномалию. Например, при трисомии 18-ой хромосомы (б-знь. Эдвардса) -это долихоцефалия (89,6%), флексорное положение кистей (96,1%), "стопа-качалка" (76,2%), короткий и широкий пальцев стопы (70,6%). При трисомии по 13-й хромосоме (б-знь. Патау) –это разщелина верхней губы и нёба (68,7%), флексорное положение кистей (44,4%), косоглазие (31,4%), дефект скальпа (30,5%) С - признаки, характерные лишь для отдельных хромосомных аномалий: крик кошки при синдроме 5р-, аллопеция при синдроме 18р-

Хромосомные аномалии Если хромосомная аномалия присутствует в половой клетке, то все клетки будущего организма наследуют эту аномалию (что приводит к развитию полной формы наследственной болезни). Если хромосомная аномалия присутствует в соматической клетке, то только часть клеток организма имеет хромосомную аномалию (хромосомный мозаицизм).
. Нерасхождение")
Нерасхождение хромосом в мейозе дает нарушение во всех клетках потомка (генеративная мутация). Нерасхождение при митозе дает нарушение только в потомстве данной клетки (соматическая мутация). Такой организм называется «мозаик».

Мозаицизм - сочетание клеток с нормальными и аномальными хромосомами. Часто встречаются родители, внешне вполне нормальные, которые имеют детей с аномальными половыми хромосомами. Организм, у которого в результате соматической мутации (хромосомной или генной) часть клеток стала генетически отличаться от остальных клеток. Хромосомных мозаиков иногда называют миксоплоидами. Мозаик может возникнуть вследствие митотического нерасхождения или в результате утери хромосомы вследствие анафазного отставания Обследование клеток крови, костного мозга и кожи
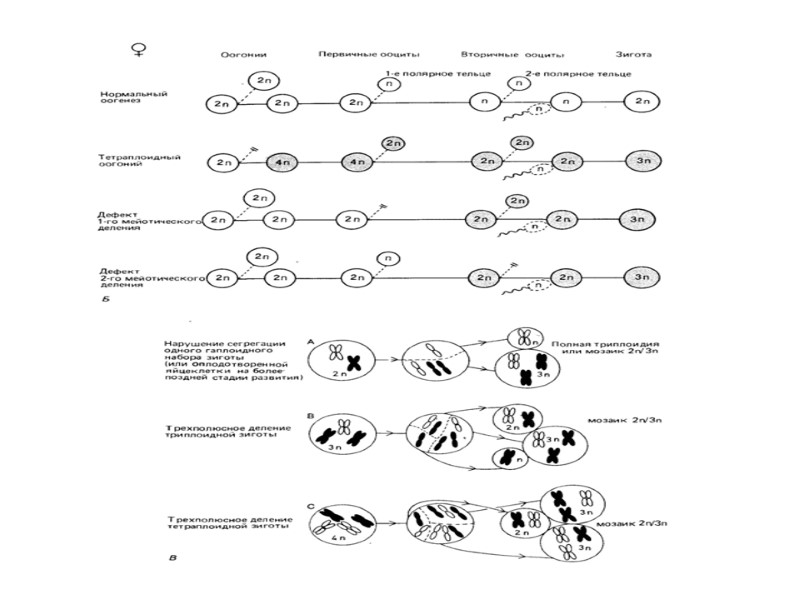

Нарушение плоидности Наследственные болезни Геномные мутации - изменения количества хромосом в геноме Анеуплоидии – изменение числа хромосом, не кратное гаплоидному набору Полиплоидии – изменение количества хромосом, кратное гаплоидному набору (3n ,4n) k = 1 - гаплоидия k = 2 – норма k = 3 - триплоидия k = 4 - тетраплоидия и так далее 2n + 1 - трисомия 2n + 2 - тетрасомия 2n – 1- моносомия 2n – 2 - нулисомия

Полиплоидия у растений приводит к увеличению размеров всех частей тела

У животных и человека приводит к гибели плода
 характер нарушения зависит от того, чьих хромосомных набора два, а чьих")
При триплоидии (3n) характер нарушения зависит от того, чьих хромосомных набора два, а чьих один 2 от матери + 1 от отца – плод выглядит нормально, но плацента недоразвита 2 набора от отца + 1 от матери – маленький плод, но очень большая плацента, возможен пузырный занос Пузырный занос это продукт зачатия, при котором не происходит нормального развития эмбриона, а ворсины плаценты разрастаются в виде пузырей, наполненных жидкостью.

Анеуплоидии – изменение количества отдельных хромосом Абсолютное большинство эмбрионов с анеуплоидией погибает на ранних сроках беременности. Чем меньше генов в хромосоме, тем вероятнее, что плод с анеуплоидией доживет до рождения. Нарушения развития всегда затрагивают многие органы и ткани

Примерное количество генов в хромосомах человека
 –")
Трисомия (от греч. tri-, в сложных словах - три и soma - тело) – наличие в хромосомном наборе диплоидного организма одной или нескольких лишних хромосом, не гомологичных друг другу. Трисомия возникает как результат нерасхождения хромосом при делении клетки.
,")
Синдром Дауна Синдром возникает из-за процесса нерасхождения хромосом при образовании гамет (яйцеклеток и сперматозоидов), в результате чего ребенок получает от матери (в 90% случаев) или от отца (в 10% случаев) лишнюю 21-ю хромосому. У большинства больных синдромом Дауна имеется три 21-х хромосомы вместо положенных двух; в 5 8% случаев аномалия связана с присутствием не целой лишней хромосомы, а ее фрагментов.
 Зависимость частоты рождения больных детей от возраста матери")
Синдром Дауна (трисомия по 21 хромосоме) Зависимость частоты рождения больных детей от возраста матери (синдром Дауна)

Признаки синдрома Дауна Плоское лицо с раскосыми глазами, Широкие губы, широкий плоский язык с глубокой продольной бороздой на нем Голова круглая, скошенный узкий лоб, ушные раковины уменьшены в вертикальном направлении, с приросшей мочкой, глаза с пятнистой радужной оболочкой (пятна Брушфельда - Brushfield's spots). Волосы на голове мягкие, редкие, прямые с низкой линией роста на шее. Укорочение и расширение кистей и стоп (акромикрия). Мизинец укорочен и искривлен, на нем только две сгибательные борозды. На ладонях только одна поперечная борозда (четырехпалая). Отмечаются неправильный рост зубов, высокое небо, Изменения со стороны внутренних органов, особенно пищевого канала и сердца (50% пороки сердца).

Синдром Дауна- трисомия 21

Диагностика синдрома Дауна Разработаны новые диагностические тест-системы для беременных, которые позволяют достаточно быстро обнаружить синдром Дауна у плода в первом триместре и ультразвуковое исследование. В анализе крови определяют так называемый плазменный белок A и гормон под названием хорионический гонадотропин человека. С помощью ультразвука обнаруживают характерные для синдрома Дауна кожные складки на задней поверхности шеи.

Мир равных возможностей Наследственные болезни Синдром Дауна – не трагедия, если тебя любят! 21 марта – Международный день человека с синдромом Дауна

Синдром Патау, трисомия 13

Трисомия 13 – синдром Патау

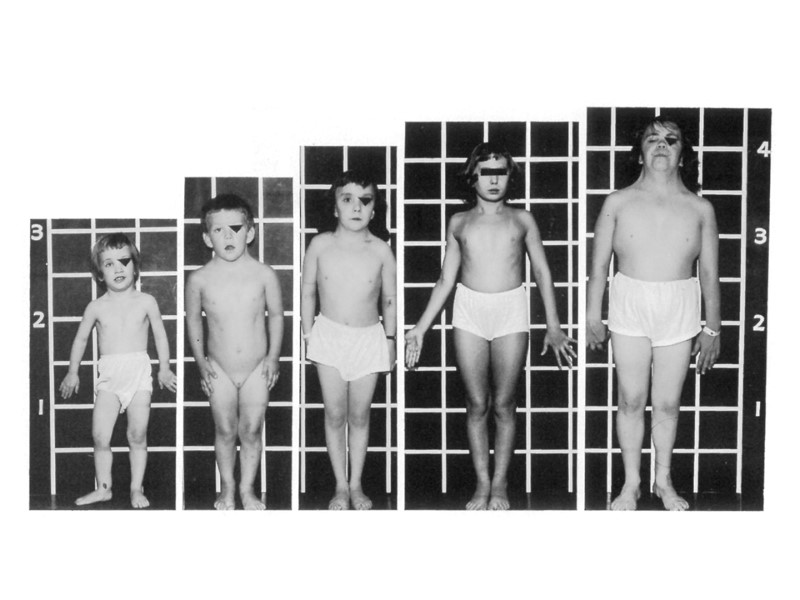
Синдром Патау Встречается с частотой 1:7000-1:14000. Имеются два цитогенетических варианта синдрома Патау: простая трисомия и робертсоновская транслокация. Характерным осложнением беременности при вынашивании плода с синдромом Патау является многоводие: оно встречается почти в 50 % случаев Синдрома Патау. При синдроме Патау наблюдаются тяжелые врожденные пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорожденных встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезенки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития. В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95 % — до 1 года). Однако некоторые больные живут в течение нескольких лет. Более того, в развитых странах отмечаются тенденция увеличения продолжительности жизни больных синдромом Патау до 5 лет (около 15 % детей) и даже до 10 лет (2 — 3 % детей). Оставшиеся в живых страдают глубокой идиотией.

Симптомы синдрома Патау Многоводие у беременной в 50% случаев. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). Микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорожденных встречаются пороки развития сердца. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезенки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития. В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95 % — до 1 года). Однако некоторые больные живут в течение нескольких лет.

Полидактилия нижних конечностей

Полидактилия верхних конечностей

Новорожденный мальчик с диагностированным синдромом Патау (разщелина верхней губы и нёба, полидактилия левой кисти, дефект межпередсердной перегородки) Мальчик 7 лет с синдромом Патау (врожденная глухота и слепота)

Трисомия 18 – синдром Эдвардса

Синдром Эдвардса, трисомия 18 Стопа-качалка Кисты в головном мозге
 Стопа-качалка Гипогенитализм у мальчика")
Синдром ЭДВАРДСА(18+) Стопа-качалка Гипогенитализм у мальчика

Синдром Эдвардса Популяционная частота примерно 1:7000. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков. В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма. Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.

Циклопия с proboscis

Симптомы синдрома Эдвардса Дети с трисомией 18 рождаются с низким, в среднем 2177 г, весом. При этом длительность беременности — нормальная или даже превышает норму. Аномалии мозгового и лицевого черепа. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщен и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса Продолжительность жизни детей : 60 % детей умирают в возрасте до 3 мес, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены..

Синдром Эдвардса Наследственные болезни Кариотип человека с синдром трисомии 18

Синдром Варкани ТРИСОМИЯ 8

Трисомия 8 Цитогенетически констатирован мозаицизм по хромосоме из группы С или D. Полные трисомии 8, как правило, летальны. Встречается с частотой не более чем 1:5000, преобладают больные мальчики (соотношение мальчиков и девочек 5:2). Большинство описанных случаев (около 90%) относится к мозаичным формам. Для болезни наиболее характерны отклонения в строении лица, пороки опорно-двигательного аппарата и мочевой системы. При клиническом обследовании выявляются выступающий лоб, косоглазие, эпикантус, глубоко посаженные глаза, высокое нёбо (иногда расщелина), толстые губы, вывернутая нижняя губа, большие ушные раковины. При УЗИ выявляются аномалии позвоночника (добавочные позвонки, неполное закрытие позвоночного канала), аномалии формы и положения ребер или добавочные ребра. Со временем у больных проявляются умственная отсталость, гидроцефалия, паховая грыжа, новые контрактуры, аплазия мозолистого тела, новые изменения скелета (кифоз, сколиоз, аномалии тазобедренного сустава, узкий таз, узкие плечи).

Синдром трисомии хромосомы 9. Нерасхождение хромосом на ранних стадиях бластулы. Цитогенетически отмечены полные формы трисомии и мозаики (50%). Микроцефалия, глубокопосаженные глаза, высокий лоб, широкая переносица, бульбообразный нос, высокое небо, часто с расщелиной. Ушные раковины деформированы и низко посажены, отмечается короткая шея с низкой линией роста волос. Наблюдаются аномалии развития опорно-двигательного аппарата, включающие дисплазию тазобедренного сустава, вывих локтевого или коленного суставов, аномалии ребер. У мальчиков выражен крипторхизм и микропенис. Патология сердечно-сосудистой системы, почек, комплексные пороки желудочно-кишечного тракта. Прогноз жизни неблагоприятный. Большинство больных погибают в первые 4 месяца жизни, особенно от респираторных инфекций.

Синдром трисомии хромосомы 14. Основными цитогенетическими формами являются мозаики. Кроме того, часто встречаются транслокационные варианты, включающие робертсоновские транслокации 14/14. Основными диагностическими признаками синдрома являются: микроцефалия, асимметрия лица, высокий и выступающийлоб, нос короткий и бульбообразный, губы полные, высокое небо, часто с расщелинами. Ушные раковины низко посажены, с маленькими мочками. Короткая шея, узкая и деформированная грудная клетка, крипторхизм, гипогонадизм и маленький пенис. Из пороков внутренних органов выражены пороки сердечно-сосудистой системы, смещение почки и почечная недостаточность, астма и дерматозы. Прогноз жизни неблагоприятный, однако отмечены больные в возрасте 13,5 лет.

Образование гамет ♀ Вследствие нерасхождения Х-хромосом у женского организма могут возникнуть гаметы с двумя Х-хромосомами: 22 + ХХ и без Х-хромосом: 22 + 0

При оплодотворении яйцеклеток нормальными сперматозоидами Возможно образование следующих зигот: 22 + ХХ + 22 + У = 44 + ХХУ (синдром Клайнфельтера ) 22 + ХХ + 22 + Х = 44 + ХХХ (трисомия X-хромосомы) 22 + 0 + 22 + Х = 44 + 0Х (моносомия X-хромосомы, синдром Тернера) 22 + 0 + 22 + У = 44 + 0У (не были найдены – не выживают)

Образование гамет ♂ Вследствие нерасхождения Х и У-хромосом у мужского организма могут возникнуть гаметы: 22 + ХУ 22 + 0

При оплодотворении нормальных яйцеклеток сперматозоидами Возможно образование следующих зигот: 22 + Х + 22 + ХУ = 44 + ХХУ (синдром Клайнфельтера ) 22 + Х + 22 + 0 = 44 + 0Х (моносомия X-хромосомы, синдром Тернера)

Трисомии по половым хромосомам Синдром Клайнфельтера - трисомия по Х хромосоме (47,XXY, ХХХУ, ХУУ и т.д.). Встречается с частотой 1:500-1:750. Синдромы три – и полисомии по X хромосоме - 47,ХХX (1 : 1000 - 2000 ); 48,ХХХХ; 49,ХХХХХ (редко). Синдром дисомии по Y-хромосоме (47,ХYY) (1:800). Наследственные болезни

Аномалии половых хромосом

Синдром Клайнфельтера характеризуются наличием как минимум одной лишней X хромосомой у мальчиков, что приводит к нарушению полового созревания у них. Клинически впервые описан Клайнфельтером в 1942 г. Популяционная частота составляет 1 : 1000 лиц мужского пола. Синдром Клайнфельтера встречается примерно у 1/800 родившихся живыми мальчиков. Лишнюю Х хромосому ребенок получает от матери в 60 % случаев.

Синдром Клайнфельтера, 47, XXY При рождении синдром Клайнфельтера клинически не проявляется. Первые отчетливые фенотипические признаки заболевания появляются в пре- и пубертатном периодах онтогенеза. Высокий рост, непропорционально длинные конечности Жироотложение по женскому типу Гипоплазия семенников, гипогенитализм, вторичные половые признаки развиты плохо, оволосение по женскому типу, гинекомастия (50%), бесплодие Брахицефалия, низкий рост волос на затылке, клинодактилия V-ого пальца, сколиоз, радиоульнарный синостоз Судороги, атаксия, тремор нарушены слуховое восприятие и обработка информации, а также навыки чтения Клиническая вариабельность значительная, многие мальчики и мужчины с кариотипом 47, XXY имеют обычную внешность и нормальный интеллект. Предрасположенность к асоциальному поведению

В большинстве случаев неправильное расхождение половых хромосом происходит в гаметах родителей. Имеют место и мозаичные варианты, например 47, XXY/46, XY. Наличие лишней X хромосомы приводит к аплазии эпителия тестикул, которые в дальнейшем гиалинизируются. Это приводит у взрослых больных к азооспермии и бесплодию. В 15 % случаев наблюдается мозаицизм. Эти мужчины могут иметь детей. У некоторых мужчин может быть 3,4 и даже 5 Х-хромосом вместе с одной Y-хромосомой. С увеличением числа Х-хромосом возрастает тяжесть умственной отсталости и пороков развития.

Синдром Клайнфельтера Цитогенетический анализ, кариотипирование часто отмечается повышенная экскреция с мочой фолликулостимулирующего гормона

Анеуплоидии по половым хромосомам не приводят к тяжелым нарушениям развития благодаря способности Х хромосомы образовывать тельце Барра

Формы анеуплоидий Моносомия — наличие в генотипе всего одной из пары гомологичных хромосом. Моносомия по половой хромосоме - синдром Шерешевского –Тернера (генотип X0, пол — женский). Популяционная частота 1:3000 новорожденных. Ребенок с синдромом Шерешевского-Тернера Наследственные болезни

Синдром ШЕРЕШЕВСКОГО-ТЕРНЕРА Крыловидная Лимфатический отёк складка на шее стопы

Клинические признаки синдрома Шерешевского-Тернера 45,ХО Низкий рост Своеобразная “щитоподобная” грудная клетка Широко расставлены соски (90%) Крыловидные складки на шее Деформорованные ушные раковины (80%) Вальгусная деформация локтей, короткая IV пястная кость Остеопороз Пигментные пятна на коже
 Низкий рост (98%), крыловидные кожные складки на")
ХРОМОСОМЫ X МОНОСОМИИ С-М (с-м ШЕРЕШЕВСКОГО-ТЕРНЕРА) Низкий рост (98%), крыловидные кожные складки на шее (56%), широкая грудная клетка (60%), Х-подобное искривление голеней (56%) Половой инфантилизм (94%), первичная аменорея (96%), бесплодие (99%) У новорожденных периферический лимфатический отёк (40%) Короткая шея (71%), низкая линия роста волос (73%), гипоплазия или гипертрофия ногтевых пластинок (73%), гиперпигментация кожи (60%), микрогнатия (40%) Гипертензия (27%), аномалии мочевой системы (38%), снижение слуха (52%), в 16% - снижение умственного развития

Врожденная аномалия матки и яичников

Пренатальная диагностика синдрома Шерешевского-Тернера Пузырчатая гигрома шейного отдела плода и отёк плода
")
СИНДРОМ ШЕРЕШЕВСКОГО —ТЕРНЕРА (45, ХО)

Синдром диплосомии-Y 47, XYY Частота синдрома по среднестатистическим данным составляет среди новорожденных около 1:1000. Иногда приводятся значительно более высокие данные— 1:250. Наиболее частым признаком является высокий рост, который у взрослых больных составляет в среднем 186 см. Однако этот признак не является абсолютным, так как в литературе имеются описания мужчин с кариотипом 47, XYY среднего роста. У части больных отмечаются нерезко выраженные евнухоидные черты телосложения и диспластические признаки: неправильное строение зубов, увеличение нижней челюсти, аномальный прикус, девиация коленных и локтевых суставов, радиоульнарный синостоз. У некоторых больных обнаруживается повышение уровня андрогенов и лютеинизирующего гормона. Половая функция не нарушена. Наличие добавочной Y-хромосомы может и не сопровождаться клинической патологией, но, несомненно, оно коррелирует как с интеллектуальным недоразвитием, так и с эмоционально-волевыми нарушениями. Не исключены некоторые особенности поведения таких лиц: при соответствующих условиях они склонны к агрессивным и даже криминальным поступкам. При цитогенетическом исследовании с помощью люминесцентной микроскопии в буккальных мазках обнаруживается Y-хроматин. При анализе кариотипа выявляется дополнительная Y-хромосома.

Трисомия Х, 47ХХХ Частота трисомии-Х составляет среди новорожденных девочек и женщин 1:1000, среди умственно отсталых — 0,59 %. Большинство девочек и женщин с трисомией-Х выявлены среди больных психиатрических больниц. Клинические проявления весьма полиморфны, а у части пациентов с трисомией-Х вообще не обнаруживается каких-либо отклонений в физическом и психическом развитии. Умственная отсталость, которая отмечается у 75 % больных. Особое внимание привлекает частота заболевания шизофренией. У многих больных с трисомией-Х наблюдаются задержка физического развития, негрубые диспластические признаки: эпикант, высокое твердое небо, клинодактилия мизинцев. Реже встречаются больные высокого роста. У некоторых пациентов отмечается бесплодие, обусловленное недоразвитием фолликулов. Диагноз ставят только при цитогенетическом исследовании: выявляют 47 хромосом и двойной половой хроматин. Описано также много случаев так называемой полисомии-Х: тетрасомия (ХХХХ) и пентасомия (ХХХХХ) с соответствующим увеличением количества телец полового хроматина. В этих случаях степень психического недоразвития выражена более и коррелирует с количеством дополнительных Х-хромосом.
 –")
Нуллисомия (от лат. nullus - никакой, несуществующий и греч. sōma - тело) – тип геномной мутации, заключающийся в отсутствии в клетках организма какой-либо пары хромосом, в норме присущей данному виду. Организмы с нуллисомией называются нуллисомиками. Нуллисомия, в особенности у высших животных, обычно ведёт к гибели организма.

Хромосомные мутации часто являются результатом нарушения кроссинговера – обмена участками между хромосомами, происходящего в профазе 1 мейоза внутрихромосомные межхромосомные
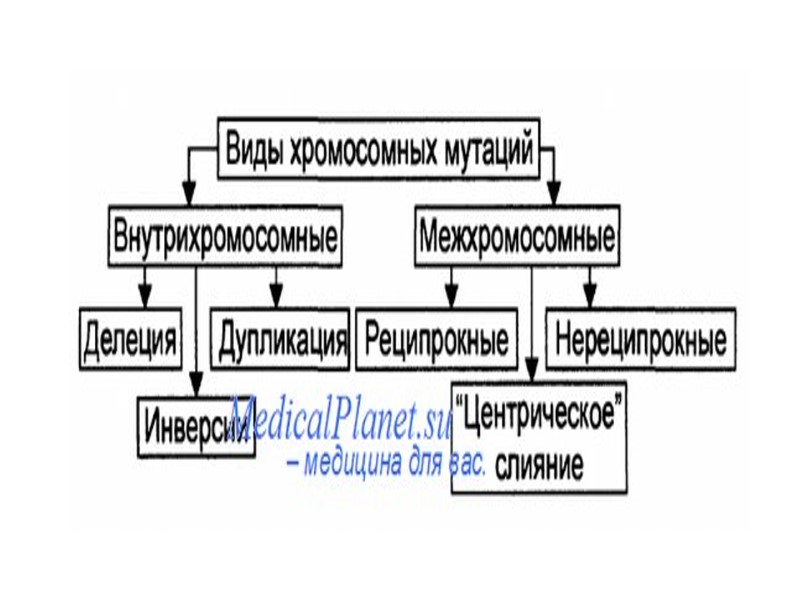
 Дупликация(dup) Инверсия(inv)")
Делеция(del) Дупликация(dup) Инверсия(inv)
 – тип хромосомной перестройки, при которой из ДНК")
ДЕЛЕЦИЯ (от лат. deletio – уничтожение) – тип хромосомной перестройки, при которой из ДНК выпадает участок генетического материала. Делеция может быть следствием разрыва хромосомы или результатом неравного кроссинговера. Делеции подразделяют на: интерстициальные (потеря внутрен него участка) терминальные (потеря концевого участка).

Инверсия —поворот участка хромосомы на 180°. Инверсии являются сбалансированными внутрихромосомными перестройками. Различают парацентрические (инвертированный фрагмент лежит по одну сторону от центромеры) и перицентрические (центромера находится внутри инвертированного фрагмента) инверсии. Инверсии, как правило, не влияют на фенотип носителя. Гетерозигота по инверсии может иметь сниженную фертильность и вероятность рождения потомства с аномальным фенотипом из-за образования аберрантных рекомбинантных хромосом в мейозе.

Инверсия в А-плече политенной хромосомы комара-звонца, род Axarus

Варианты инверсий перицентрическая парацентрическая

Дупликации Дупликации представляют собой класс перестроек, который объединяет как внутри- , так и межхромосомные перестройки. Дупликация — это появление дополнительной копии участка хромосомы, которая может располагаться сразу за тем районом, который дуплицирован, тогда это тандемная дупликация, либо в новом месте или в другой хромосоме. Новая копия может образовать отдельную маленькую хромосому со своими собственными теломерами и центромерой, тогда это свободная. Тандемные дупликации появляются в половых клетках при мейозе в результате неравного кроссинговера (в этом случае второй гомолог несет делецию) или в соматических клетках в результате неаллельной гомологичной рекомбинации при репарации двунитевого разрыва ДНК. В процессе кроссинговера у гетерозиготы при конъюгации хромосомы с тандемной дупликацией и нормальной хромосомы, как и при делеции, формируется компенсационная петля.
 при неправильном разделении хроматид p q p q p q p")
Возникновение изохромосомы (i) при неправильном разделении хроматид p q p q p q p q Нормальное расхождение в анафазе Изохромосомы (isochromosome) – аномальная хромосома, которая образуется на стадии анафазы деления клеток, когда центромер делится горизонтально, а не продольно. В результате образуются одна хромосома с двумя длинными плечами и одна с двумя короткими. Изохромосомные аберрации — образование одинаковых, но зеркальных фрагментов двух разных хромосом, содержащих одни и те же наборы генов. Это происходит в результате поперечного разрыва хроматид через центромеры (отсюда другое название — центрическое соединение).
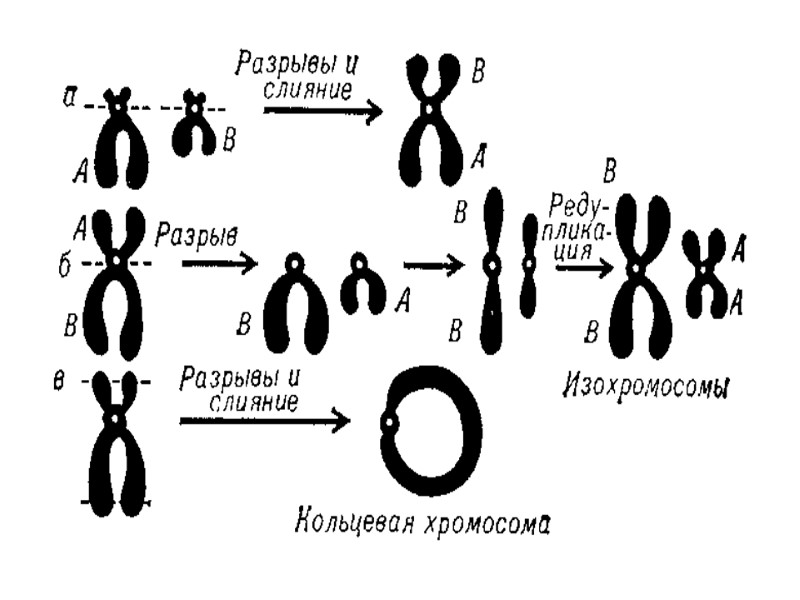
 кольцо ацентрические фрагменты")
Образование кольцевой хромосомы (r) кольцо ацентрические фрагменты

Межхромосомные перестройки - транслокации Взаимные Невзаимные Робертсоновские Межхромосомные аберрации — обмен фрагментами между негомологичными хромосомами. Они получили название транслокаций. Различают три варианта транслокаций: реципрокные (обмен фрагментами двух хромосом), нереципрокные (перенос фрагмента одной хромосомы на другую), робертсоновские (соединение двух акроцентрических хромосом в районе их центромер с потерей коротких плеч, в результате образуется одна метацентри-ческая хромосома вместо двух акроцентрических).

Транслока́ция —перенос участка хромосомы на негомологичную хромосому. Реципрокные транслокации -взаимный обмен участками между хромосомами. Робертсоновские транслокации, или центрические слияния - слияние акроцентрических хромосом с полной или частичной утратой материала коротких плеч. Различные транслокации в соматических клетках приводят к развитию лимфом, сарком, лейкозов.
:1 – невзаимная (инсерция), 2 – взаимная (реципрокная)")
Транслокация (t):1 – невзаимная (инсерция), 2 – взаимная (реципрокная)

Реципрокные транслокации Реципрокные транслокации являются сбалансированной хромосомной перестройкой, при их формировании не происходит потери генетического материала. Они являются одной из самых распространенных хромосомных аномалий в человеческой популяции, частота носительства варьирует от 1/1300 до 1/700. Носители реципрокных транслокаций, как правило, фенотипически нормальны, при этом имеют повышенную вероятность бесплодия, сниженной фертильности, спонтанных выкидышей и рождения детей с врождёнными наследственными заболеваниями, так как половина гамет у них генетически несбалансирована из-за неравновесного расхождения перестроенных хромосом в мейозе. Врождённые сбалансированные транслокации могут давать аномальный фенотип в случае, если точка разрыва находится внутри гена или внутри его регуляторных последовательностей, а также если перестройка приводит к изменению экспрессии гена за счёт так называемого эффекта положения.

Филадельфийская хромосома Первой описанной структурной геномной перестройкой в соматических клетках, которая вызывает онкологическое заболевание, является так называемая филадельфийская хромосома — Ph. Сейчас известно, что филадельфийская хромосома возникает вследствие реципрокной транслокации между хромосомами 9 и 22, и эта мутация вызывает 95 % случаев хронического миелобластного лейкоза. Также эта мутация является одной из самых распространённых при В-клеточном остром лимфобластном лейкозе взрослых В результате транслокации t(9;22)(q34;q11) ген ABL1 из хромосомы 9 объединяется с геном BCR хромосомы 22. Активность нового химерного белка приводит к нечувствительности клетки к воздействию факторов роста и вызывает её безудержное деление.

Детекция филадельфийской хромосомы при помощи флуоресцентной гибридизации in situ
 (центрическое слияние) Между разными акроцентриками – у человека 13,14,15,21,22 Кариотип при")
Робертсоновская транслокация (rob) (центрическое слияние) Между разными акроцентриками – у человека 13,14,15,21,22 Кариотип при «семейном» синдроме Дауна

Робертсоновские транслокации По некоторым данным, их частота составляет 1:1000 новорожденных. Их носители фенотипически нормальны, однако у них существует риск самопроизвольных выкидышей и рождения детей с несбалансированным кариотипом. Большинство Робертсоновских транслокаций затрагивают хромосомы 13 и 14. В структуре обращаемости на пренатальную диагностику лидерами оказываются носители der(13;14) и der(14;21). Последний случай, а именно, Робертсоновская транслокация с участием хромосомы 21 приводит к так называемому «семейному» (наследуемому) синдрому Дауна. Показано, что два плеча 2-й хромосомы человека соответствуют 12 и 13 хромосомам шимпанзе. Возможно, 2-я хромосома образовалась в результате робертсоновской транслокации двух хромосом обезьяноподобного предка человека.
 хромосомы и парные ацентрические фрагменты")
В результате транслокаций могут возникать дицентрические (dic) хромосомы и парные ацентрические фрагменты
 — женщина с делецией короткого плеча хромосомы")
Номенклатура хромосомных мутаций. del Делеция, например 46,XX,del(5p) — женщина с делецией короткого плеча хромосомы 5 dup Дупликация, например 46,XY,dup(ll)(ql2) — мужской кариотип с дупликацией сегмента ql2 хромосомы 11 fra Ломкий сайт i Изохромосома, например 46,X,i(Xq) — женский кариотип, одна из хромосом Х представлена изохромосомой по длинному плечу inv Инверсия, например 46,XY,inv(10)(pl3ql2) — мужской кариотип, перицентрическая инверсия с точками разрыва р13 и ql2 rob Робертсоновская транслокация, например, 45,XX,rob (14q21q) — женщина со сбалансированной робертсоновской транслокацией длинных плеч хромосом 14 и 21, или 46,XX,-14, r Кольцевая хромосома, например 46,ХХ, г(16) — женщина с кольцевой хромосомой 16 t Транслокация, например 46,ХХ, t(2;4)(q21;q21) — женщина с реципрокной трансплантацией, включающей длинное плечо хромосомы 2, начиная с сегмента 2q21, и длинное плечо хромосомы 4, начиная с сегмента 4q21

Кроме того, хромосомные мутации бывают: Спонтанные/индуцированные Соматические /генеративные Вредные/полезные/нейтральные

С клинической точки зрения хромосомные мутации удобнее делить на Сбалансированные (нет потери или добавления генов) Например, инверсия, реципрокная транслокация Несбалансированные (гены теряются или добавляются) Например, делеция дупликация

Синдром Вольфа-Хиршхорна

Частота этого синдрома низкая — 1 : 50000 рождений. Средняя продолжительность жизни примерно до 30 лет. Делеция короткого плеча 4-й хромосомы. Наряду с делецией хромосом, патология у новорождённых может быть обусловлена инверсиями, дупликациями, изохромосомами. задержкой физиологического, умственного и психомоторного развития. Также могут проявляться в большинстве случаев тяжелейшие пороки сердца, почек. У новорождённых небольшой вес при нормальной продолжительности беременности (до 2 кг). Среди внешних признаков могут отмечаться: микроцефалия, клювовидный нос, эпикант, антимонголоидный разрез глаз (опущение наружных углов глазных щелей), аномальные ушные раковины, расщелина верхней губы и нёба, маленький рот, деформация стоп и др.

Синдром "кошачьего крика"

Нарушение целостности хотя бы одной хромосомы приводит к тяжёлым последствиям. Синдром «кошачьего крика» связан с делецией короткого плеча 5-ой хромосомы.

Причины Частота проявления 5p- 1:30000- 1:50000 М 1: Ж 1,3
 гипотония")
Характерные особенности специфический плач низкая масса тела (до 2500 кг) гипотония задержка психомоторного развития соматические заболевания

Фенотип лунообразное лицо гипертелоризм антимонголоидный разрез глаз эпикантус возможен птоз несколько уплощенный нос низко расположенные ушные раковины микроцефалия маленькая нижняя челюсть короткая шея с крыловидными складками

Зрение и слух Зрение: миопия косоглазие астигматизм нистагм Слух: гиперакузия
 повышенная импульсивность (90%) агрессия самоагрессия стереотипии негативизм")
Особенности поведения гиперактивность (25 – 80%) повышенная импульсивность (90%) агрессия самоагрессия стереотипии негативизм
. Цитогенетические варианты могут быть различны: частичная делеция короткого плеча 9")
Синдром Альфи (синдром 9р-). Цитогенетические варианты могут быть различны: частичная делеция короткого плеча 9 хромосомы, изохромосома 9q, несбалансированные транслокации, однако во всех случаях наблюдается потеря сегмента 9р22. Диагностическими признаками заболевания являются: тригоноцефалия, резковыступающий лоб, монголоидный разрез глаз, эпикант, экзофтальм, гипертелоризм, уплощенная и широкая переносица, маленький рот с большой верхней губой, высокое небо. Ушные раковины без мочки или она недоразвита, со сглаженным завитком. Шея короткая, отмечен гипертелоризм сосков. Пальцы рук и ног длинные, с дополнительными сгибательнымискладками; ногти широкие, выпуклые, квадратной формы. У девочек выражена гипоплазия малых и больших половых губ, у мальчиков - гипоплазия мошонки и полового члена. Из пороков внутренних органов отмечено поражение сердечно-сосудистой системы и гидронефроз почек. Умственная отсталость в стадии имбецильности, реже – дебильности.

Ребёнок 3 лет Женщина 21 года Синдром частичной трисомии по короткому плечу хромосомы 9 (9р+)

Микроцефалия, брахицефалия; Гипертелоризм, антимонголоидний розрез глаз, енофтальм; Низко расположенные ушные раковины, короткая шея; Кифоз, сколиоз, гипоплазия терминальных фаланг пальцев, синдактилия ІІ-III на стопах і ІІ-ІV на руках; Задержка полового созревания, у 25% - пороки сердца и почек. Синдром частичной трисомии по короткому плечу хромосомы 9 (9р+)

Микроцефалия, большой лоб, плоское круглое лицо; Гипертелоризм, антимонголоидный разрез глаз; Маленький нос с запавшей переносицей, дугоподобный рот с виступающей верхней губой, деформованные ушные раковины; Короткая шея, синдактилия кистей и стоп, сандалеподобная щель; Выраженая гипотрофия, пороки сердца и почек; Синдром частичной трисомии по короткому плечу хромосомы 10 (10q+)
 Долихоцефалия, диспроhорция лицевой и мозгового")
Синдром частичной трисомии по короткому плечу хромосомы 10 (10p+) Долихоцефалия, диспроhорция лицевой и мозгового отделов черепа; Микрофтальмия, нистагм, микрокорнеа, колобома зорового нерва; Треугольный рот, аномалии зубов; Сколиоз, флексорная деформация суставов рук, гипотония и гипоплазия скелетных мышц; Отставание в психомоторного развития, гипотрофия.
 Резко выраженая гипоплазия; Микроцефалия, короткий")
Синдром частичной трисомии по короткому плечу хромосомы 10 (11q+) Резко выраженая гипоплазия; Микроцефалия, короткий нос, втянута нижняя губа; Гипертонус конечностей у новорожденных, вывих бедра, косолапость; Порок сердца, почек, дефекты нервной трубки.
 Задержка роста; Тригоноцефалия, гипертелоризм, епикант; Клинодактилия, аплазия дистальных")
ХРОМОСОМЫ 11р- (делеции короткого плеча11-ой хромосомы) Задержка роста; Тригоноцефалия, гипертелоризм, епикант; Клинодактилия, аплазия дистальных фаланг пальцев; Врожденные пороки сердца, удвоение почек, пилоростеноз.
 Плоский профиль лица, сплощенная спинка")
ХРОМОСОМЫ 18q- (делеции длинного плеча18-ойхромосомы, кольцевой хромосомы 18) Плоский профиль лица, сплощенная спинка носа, гипертелоризм, глубоко посаженные глаза, "карпячий рот", тонкая верхняя губа; Сужение внешних слуховых каналов, деформованные ушные раковины - “уши сатира"; Нистагм, косоглазость, глаукома, епикант, косой разрез глазных щелей, атрофия зрительных нервов; Руки длинные с конусовидными пальцами, косолапость; В 100% умственная отсталость; Мышечная гипотония, гипоплазия половых органов, крипторхизм.

ХРОМОСОМЫ 18р- Низкая маса тела при рождении, задержка роста, укороченные конечности; Гипертелоризм, епикант, птоз, широкий рот с опущеными уголками губ, кариес, больщие оттопыренные уши, короткая шея, широкая вдавленая грудная клетка, синдактилия стоп; Реже - ариненцефалия, цебоцефалия, циклопия, "волчья пастья", "зайчья губа", катаракта, страбизм, отставание в речевом развитии.

Синдром кольцевой хромосомы 22. Цитогенетические варианты могут быть различны: кольцевая хромосома 22, делеции длинного плеча этой хромосомы и мозаичные варианты. В 90% случаев данные хромосомные нарушения являются мутациями de novo. Основными диагностическими признаками заболеванияя вляются: микроцефалия, эпикант, гипертелоризм, крупные выступающие глаза ("глаза лани"), расщелина язычка инеба, густые брови и ресницы. Кроме того, отмечается дисплазия тазобедренных суставов, клинодактилия и синдактилия. Пороки внутренних органов не характерны. Умственная отсталость проявляется как легкой, так и выраженной олигофренией. Больные легко возбудимы, отмечается частая смена настроения, нарушение координации, а также грубое недоразвитие речи, вплоть до ее отсутствия.
 Низкая масса тела при рождении,")
ХРОМОСОМА 21q- (делеции длинного плеча 21-ой хромосомы) Низкая масса тела при рождении, черепно-лицевые дисморфии (микроцефалия, епикант, антимонголоидный разрез глаз, микрогнатия); Сколиоз, клинодактилия, косолапость, мышечная гипертония, крипторхизм; В 30% - аномалии глас, пороки почек; Діти резко отстают в психомоторном развитии.

 Выраженая пренатальная гипотрофия и дальнейшая задержка психомоторного развития; Микроцефалия, тригоноцефалия,")
ХРОМОСОМА 13q- (синдром ОРБЕЛИ) Выраженая пренатальная гипотрофия и дальнейшая задержка психомоторного развития; Микроцефалия, тригоноцефалия, краниостеноз; Широкое виступающая переносица, микроретрогнатия, епикант, гипертелоризм, деформированные ушные раковины; Ретинобластомы, катаракта; Гипоплазия І пальца руки, клинодактилия; У 30% аномалии мочевыделительной системы, в некоторых случаях пороки развития мозга -прозенцефалия, внутренняя гидроцефалия, гипоплазия мозжечка.
. Особый интерес к этому синдрому проявился в 1980 году,")
Синдром Лангера-Гидиона (трихо-рино-фалангиальный синдром). Особый интерес к этому синдрому проявился в 1980 году, когда была выяснена его хромосомная этиология, выражающаяся в микроделеции хромосомы длинного плеча хромосомы 8. Отмечены мозаичные формы синдрома. Основными клиническими признаками заболевания являются: тонкие и редкие волосы, бульбообразныйнос, конические эпифизы фаланг пальцев, множественные хрящевые экзостозы. Кроме этих признаков больные имеют ряд микроаномалий: широкие редкие брови, глубоко посаженые глаза, макростомию, нарушение прорезывания и расположения зубов, микрогнатию, большие и низко расположенные ушные раковины. Множественные хрящевые экзостозы проявляются до 4-х лет и располагаются везде, где есть хрящи, причем их рост усиливается в периоды активного роста организма и прекращается в возрасте 18-20 лет. У новорожденных бывает избыточная кожа. Отмечается умственная отсталость различной степени, задержка речевого развития.

Синдром Видемана-Беквита. Цитогенетически характеризует дупликацией участка короткого плеча 11 хромосомы-11р15. Основными диагностическими признаками заболевания являются: макроглоссия, макросомия с увеличением мышечной массы и подкожного жирового слоя, выступающий затылок и аномалии прикуса, связанные с гипоплазией верхней челюсти и относительной гиперплазией верхней. Характерным признаком является наличие вертикальных бороздок на мочках ушей. Описана патология развития внутренних органов: дефекты межжелудочковых перегородок, добавочная селезенка, цитомегалия коры надпочечников, незавершенный поворот кишечника. Костный возраст опережает паспортный. Психическое развитие соответствует возрасту, возможна умеренная умственная отсталость. В сегменте 11 р15 локализован ген "инсулиноподобного фактора роста II типа", при дупликации которого образуются три его копии, что приводит к появлению таких признаков синдрома как большой вес, пупочная грыжа, увеличенный язык и т.д.

Ретинобластома. Больные с ретинобластомой - злокачественной опухолью сетчатки глаза, составляют 0,6-0,8% от числавсех больных с онкозаболеваниями. Это первая опухоль, для которой установлена связь с хромосомной патологией. Цитогенетически при данном заболевании выявляется микроделеция 13 хромосомы, сегмента 13q14. Кроме микроделеции встречаются мозаичные формы и транслокационные варианты. Описано несколько случаев транслокации сегмента 13 хромосомы на Х-хромосому. Не отмечено корреляции между размерами делецированного фрагмента и фенотипическими проявлениями. Заболевание обычно начинается в возрасте около 1,5 лет и первыми признаками являются свечение зрачков, вялая реакция зрачка на свет, а затем и снижение зрения вплоть до слепоты. Осложнениями ретинобластомы являются отслойка сетчатки, вторичная глаукома. В1986 году в критическом сегменте 13q14 обнаружен ген-супрессор опухоли RB1, который явился первым антионкогеном, обнаруженным у человека.

Синдром Энгельмана (синдром "счастливой куклы"). Основными признаками заболевания являются: необычный и частый смех, специфичное лицо с гримасой улыбки, повторяющиеся кукольные стереотипные движения, отсутствие речи. Имеется выраженная умственная отсталость. Большинство больных имеют микроделецию 15q11-q13, но эта делеция всегда материнского происхождения. Обнаружены также пациенты с типичным синдромом Энгельмана безмикроделеции, у которых выявляется однородительская дисомия хромосомы 15 отцовскою происхождения.
. Заболевание с аутосомно-рецессивным типом наследования MIM 277700). Описано в")
Синдром Вернера (синдром преждевременного старения). Заболевание с аутосомно-рецессивным типом наследования MIM 277700). Описано в 1904 году. Основными диагностическими признаками являются: преждевременное поседение иоблысение, атрофия подкожной жировой клетчатки и мышечной ткани, склеродермия, катаракта, ранний атеросклероз, эндокринная патология (сахарный диабет). Характерны бесплодие, гинекомастия, аменорея, высокий голос, склонность к злокачественным новообразованиям. Больные умирают в возрасте 30-40 лет. Цитогенетически характеризуется клеточнымиклонами с разными хромосомными транслокациями (мозаицизм по различным транслокациям). Ген заболевания локализован в сегменте 8р11-р12.

Синдром Вильямса (лицо "эльфа") Популяционная частота 1 на 10000. Выделяют 2 группы больныхс данным синдромом: 1) классическая форма с делецией 7q11, которая обнаруживается в 96% случаев; 2) более редкая форма, при которой обнаруживаются делеции в 11 и 22 хромосомах - 11q13-q14 и 22q-, выявляемые в основном с помощью молекулярно-цитогенетических методов исследования. Основными диагностическими признаками синдрома являются: необычное лицо, эпикант, отечность век, короткий нос с открытыми вперед ноздрями, полные щеки, микрогения. Патология внутренних органов включает надклапанный стеноз аорты, дефекты перегородок сердца, стеноз легочной артерии. Умственная отсталость различной степени, разнообразные психические нарушения, низкий интеллект. С возрастом заболевание утяжеляется.
. Проявляется в видекожной сыпи с")
Порокератоз Мибелли. Заболевание с аутосомно-доминантным типом наследования (MIM 175800). Проявляется в видекожной сыпи с кратерообразно углубленными участками атрофии и центробежно распространяющимися кожнымилоскутами, окруженными узкими роговыми гребнями. Начинается в среднем возрасте и может приводить к раку кожи, особенно на конечностях. Обнаруживается высокая частота хромосомных нарушений в фибробластах кожи. Часто повреждается сегмент Зр14-р12. Птицеголовая карликовость или синдром Секеля. Заболевание с аутосомно-рецессивным типом наследования (MIM210600). Характеризуется низким весом при рождении, карликовостью, маленькой головой, клювовидным носом, большими глазами, узким лицом, умственной отсталостью, маленьким мозгом, панцитопенией и повышенной частотой хромосомных аберраций.

Синдром Блюма. Основными диагностическими признаками являются: низкий вес прирождении, задержка роста, узкое лицо с эритемой в виде бабочки, массивный нос, склонность к злокачественным новообразованиям. Умственная отсталость отмечается не во всех случаях. Цитогенетически характеризуется увеличением числа сестринских хроматидных обменов (СХО) на клетку до 120-150, хотя в норме их число непревышает 6-8 обменов на 1 клетку. Кроме того, с высокой частотой обнаруживаются хроматидные разрывы, а также дицентрики, кольца и хромосомные фрагменты. У больных обнаруживаются мутации в гене ДНК-лигазы 1, локализованномна 19 хромосоме- 19q 13.3, однако ген синдрома Блюма картирован в сегменте 15q26.1.

Синдром ломкой Х-хромосомы Разрывы хромосом или пробелы хроматид, возникающие с повышенной частотой в тех или иных конкретных хромосомных сегментах (так называемые ломкие участки или фрагильные сайты хромосом), не связаны с какими-либо заболеваниями. Синдром Мартина-Белл или синдром ломкой Х-хромосомы характеризуется ломкой (фрагильной) Х-хромосомой в сегменте Xq27.3, которая выявляется в специальных условиях культивирования клеток в среде с дефицитомфолиевой кислоты. Фрагильный сайт при этом синдроме получил обозначение FRAXA. Основными признаками заболевания являются: умственная отсталость, прогнатизм, широкое лицо с чертами акромегалии, большие оттопыренные уши, макроорхидизм в постпубертатном периоде, аутизм, гиперкинезы, плохая концентрация внимания, дефекты речи, более выраженные у детей. Отмечаются также аномалии соединительной ткани с гиперрастяжимостьюсуставов и пролапсом митрального клапана. Относительно полный спектр клинических признаков имеют только 60% мужчин с фрагильной Х-хромосомой, 10% больных не имеют лицевых аномалий, 10% имеют только умственную отсталость без других признаков, а 30% больных не имеют макроорхидизма.

Список источников. Литература 1. Гуттман Б., Гриффитс Э., Сузуки Д., Куллис Т. Генетика — Пер. с англ. О. Перфильева. — М.: ФАИР-ПРЕСС, 2004. — 448 с: ил. 2. Дубинин Н.П. Генетика и человек. Кн. для внеклассного чтения IX – X кл. М.: «Просвещение», 1978 г., 144 с. 3. Заяц Р.Г., Бутвиловский В.Э.Э Рачковская, И.В., Давыдв В.В. Общая и медицинская генетика. Лекции и задачи. - Ростов-на-Дону: Феникс, 2002 г., - 320 с. 4. http://med-dovidka.com.ua/content/view/1508 5. http://www.elkar.ru/dlja-vrachej/narushenienergoobmena/ 6. http://www.eurolab.ua/encyclopedia/505/4295/ 7. http://genetica.meduniver.com/ 8. http://hemofilia.spb.ru/news/ 9. http://il.ks.ua/tem_razdel_files/nasledstv_bolezni.htm 10. http://www.mma.ru/news/id13099 11. http://medicalplanet.su/Patfiz/59.html 12. http://www.scientific.ru/journal/grig/crimedna3.html Наследственные болезни
14170-7_topic_hromosomnye_bolezni.ppt
- Количество слайдов: 126