

Pharmacology.366.ppt
- Количество слайдов: 47
 RHS - 366
RHS - 366
 Animal tissues (heparin), Microbial (penicillin G), Human cells (urokinase),") Drug Sources Plants (cardiac glycosides) Animal tissues (heparin), Microbial (penicillin G), Human cells (urokinase), Gene technology (insulin). From poppy to morphine
Drug Sources Plants (cardiac glycosides) Animal tissues (heparin), Microbial (penicillin G), Human cells (urokinase), Gene technology (insulin). From poppy to morphine
 Drug Development 2. Clinical testing From drug synthesis to approval 1. Preclinical testing
Drug Development 2. Clinical testing From drug synthesis to approval 1. Preclinical testing
 Liquid preparations Solid dosage forms") Drug Administration (Dosage forms) Liquid preparations Solid dosage forms
Drug Administration (Dosage forms) Liquid preparations Solid dosage forms
 Parenteral (e. g. injections) Pulmonary (e. g. aerosols) Rectal or") Drug Administration (Dosage forms) Parenteral (e. g. injections) Pulmonary (e. g. aerosols) Rectal or vaginal (e. g. suppositories) Cutaneous application (e. g. powders and ointments).
Drug Administration (Dosage forms) Parenteral (e. g. injections) Pulmonary (e. g. aerosols) Rectal or vaginal (e. g. suppositories) Cutaneous application (e. g. powders and ointments).
 Possible Sites of Drug Entry From application to distribution
Possible Sites of Drug Entry From application to distribution
 Bioavailability is defined as the fraction of a given drug dose that reaches the circulation in unchanged form and becomes available for systemic distribution. The larger the presystemic elimination, the smaller is the bioavailability of an orally administered drug. The area under the blood concentration-time curve is a common measure of the extent of bioavailability for a drug given by a particular route. An I. V. dose of the drug, bioavailability is assumed to be equal to unity (100%). For a drug administered orally, bioavailability may be less than 100% for two main reasons (1) incomplete extent of absorption and (2) first-pass elimination. Various physiological factors reduce the availability of drugs prior to their entry into the systemic circulation, Such factors may include, but are not limited to: 1. Physical properties of the drug (hydrophobicity, solubility). 2. The drug formulation (immediate release, delayed release, sustained release, etc. ). 3. If the drug is administered in a fed or fasted state. 4. Gastric emptying rate. 5. Enzyme induction/inhibition by other drugs/foods. 6. Transporters: Substrate of an efflux transporter? (e. g. P-glycoprotein). 7. Health of the GI tract. 8. Individual Variation in Metabolic Differences (Age, enterohepatic circulation, diet). 9. Disease state (hepatic insufficiency, poor renal function).
Bioavailability is defined as the fraction of a given drug dose that reaches the circulation in unchanged form and becomes available for systemic distribution. The larger the presystemic elimination, the smaller is the bioavailability of an orally administered drug. The area under the blood concentration-time curve is a common measure of the extent of bioavailability for a drug given by a particular route. An I. V. dose of the drug, bioavailability is assumed to be equal to unity (100%). For a drug administered orally, bioavailability may be less than 100% for two main reasons (1) incomplete extent of absorption and (2) first-pass elimination. Various physiological factors reduce the availability of drugs prior to their entry into the systemic circulation, Such factors may include, but are not limited to: 1. Physical properties of the drug (hydrophobicity, solubility). 2. The drug formulation (immediate release, delayed release, sustained release, etc. ). 3. If the drug is administered in a fed or fasted state. 4. Gastric emptying rate. 5. Enzyme induction/inhibition by other drugs/foods. 6. Transporters: Substrate of an efflux transporter? (e. g. P-glycoprotein). 7. Health of the GI tract. 8. Individual Variation in Metabolic Differences (Age, enterohepatic circulation, diet). 9. Disease state (hepatic insufficiency, poor renal function).
 Pharmacokinetics is the effects of the body on the drug. It includes absorption, distribution, metabolism (biotransformation), and elimination. The interrelationship of the absorption, distribution, binding, metabolism, and excretion of a drug and its concentration at its sites of action.
Pharmacokinetics is the effects of the body on the drug. It includes absorption, distribution, metabolism (biotransformation), and elimination. The interrelationship of the absorption, distribution, binding, metabolism, and excretion of a drug and its concentration at its sites of action.
 Pharmacokinetics
Pharmacokinetics
 Pharmacokinetics: Absorption Characteristics of a drug (molecular size and shape, degree of ionization, relative lipid solubility of its ionized and non-ionized forms, and its binding to serum and tissue proteins). Ways by which small molecules cross cell membranes 1. Diffusion 3. Pinocytosis 2. Active transport
Pharmacokinetics: Absorption Characteristics of a drug (molecular size and shape, degree of ionization, relative lipid solubility of its ionized and non-ionized forms, and its binding to serum and tissue proteins). Ways by which small molecules cross cell membranes 1. Diffusion 3. Pinocytosis 2. Active transport
 Pharmacokinetics: Distribution Protein binding involves Albumin β-globulins Transcortin, transferrin, thyroxin-binding globulin Importance of protein binding for intensity and duration of drug effect
Pharmacokinetics: Distribution Protein binding involves Albumin β-globulins Transcortin, transferrin, thyroxin-binding globulin Importance of protein binding for intensity and duration of drug effect
 Pharmacokinetics: Drug metabolism Phase I reactions are catabolic and the products are often more chemically reactive. Often involve a mono-oxygenase system in which cytochrome P 450 plays a key role. Phase II reactions are anabolic and involve conjugation, which usually results in inactive products. Both phases decrease lipid solubility, thus increasing renal elimination. First-pass (pre-systemic) metabolism. Some drugs are extracted so well by the liver or gut wall that the amount reaching the systemic circulation is much less than the amount absorbed. This reduces bioavailability even when a drug is well absorbed from the gut (Morphine, Levodopa, Lidocaine)
Pharmacokinetics: Drug metabolism Phase I reactions are catabolic and the products are often more chemically reactive. Often involve a mono-oxygenase system in which cytochrome P 450 plays a key role. Phase II reactions are anabolic and involve conjugation, which usually results in inactive products. Both phases decrease lipid solubility, thus increasing renal elimination. First-pass (pre-systemic) metabolism. Some drugs are extracted so well by the liver or gut wall that the amount reaching the systemic circulation is much less than the amount absorbed. This reduces bioavailability even when a drug is well absorbed from the gut (Morphine, Levodopa, Lidocaine)
 is excreted") Pharmacokinetics: Drug elimination 1. Biliary excretion and enterohepatic circulation Vecuronium (muscle relaxant) is excreted mainly unchanged in bile. Rifampicin is absorbed from the gut and slowly deacetylated, retaining its biological activity. Both forms are secreted in the bile, but the deacetylated form is not reabsorbed, so eventually most of the drug leaves the body in this form in the faeces.
Pharmacokinetics: Drug elimination 1. Biliary excretion and enterohepatic circulation Vecuronium (muscle relaxant) is excreted mainly unchanged in bile. Rifampicin is absorbed from the gut and slowly deacetylated, retaining its biological activity. Both forms are secreted in the bile, but the deacetylated form is not reabsorbed, so eventually most of the drug leaves the body in this form in the faeces.
 Renal clearance (CLr) is defined") Pharmacokinetics: Drug elimination 2. By the kidney (renal clearance) Renal clearance (CLr) is defined as the volume of plasma containing the amount of substance that is removed by the kidney in unit time. Cp = plasma concentration, Cu = the urinary concentration, Vu = the rate of flow of urine,
Pharmacokinetics: Drug elimination 2. By the kidney (renal clearance) Renal clearance (CLr) is defined as the volume of plasma containing the amount of substance that is removed by the kidney in unit time. Cp = plasma concentration, Cu = the urinary concentration, Vu = the rate of flow of urine,
 It is the time required to reduce the") Pharmacokinetics: Elimination half life (t 1/2) It is the time required to reduce the plasma concentration of drug to half the initial concentration (the time for 50% elimination). t 12 is the relationship between apparent volume of distribution (Vd) and clearance (CL); Vd = amount of drug in the body / plasma concentration at zero time. t 12 = 0. 693 Vd / CL. At normal dosage, most drugs are eliminated at a rate proportionate to the plasma concentration (first-order kinetics; linear). If the plasma concentration is very high and normal metabolism is saturated, the rate of elimination may become fixed (zero-order kinetics). This change in kinetics may markedly prolong the apparent serum half-life and increase toxicity.
Pharmacokinetics: Elimination half life (t 1/2) It is the time required to reduce the plasma concentration of drug to half the initial concentration (the time for 50% elimination). t 12 is the relationship between apparent volume of distribution (Vd) and clearance (CL); Vd = amount of drug in the body / plasma concentration at zero time. t 12 = 0. 693 Vd / CL. At normal dosage, most drugs are eliminated at a rate proportionate to the plasma concentration (first-order kinetics; linear). If the plasma concentration is very high and normal metabolism is saturated, the rate of elimination may become fixed (zero-order kinetics). This change in kinetics may markedly prolong the apparent serum half-life and increase toxicity.
 Pharmacodynamics is the effects of the drug on the body. It deals with the study of the biochemical and physiological effects of drugs and their mechanisms of action. The effects of most drugs result from their interaction with receptor. Receptor: any cellular macromolecule that a drug binds to initiate its effects. Receptors are located either on the surface of cell membranes or within cells themselves. Classes of ligand-triggered cell-surface receptors. 1. Ion-Channel Receptors. 2. G-protein Coupled Receptors. 3. Tyrosine Kinase Linked Receptors. 4. Receptors with intrinsic ligand-triggered enzymatic activity in the cytosolic domain.
Pharmacodynamics is the effects of the drug on the body. It deals with the study of the biochemical and physiological effects of drugs and their mechanisms of action. The effects of most drugs result from their interaction with receptor. Receptor: any cellular macromolecule that a drug binds to initiate its effects. Receptors are located either on the surface of cell membranes or within cells themselves. Classes of ligand-triggered cell-surface receptors. 1. Ion-Channel Receptors. 2. G-protein Coupled Receptors. 3. Tyrosine Kinase Linked Receptors. 4. Receptors with intrinsic ligand-triggered enzymatic activity in the cytosolic domain.
 Pharmacodynamics Factors Governing Drug-Receptor Interactions 1. Affinity (the ability of the drug to bind to the receptor). 2. Intrinsic activity = efficacy (the ability the drug to produce a detectable effect). Agonists have both affinity as well as intrinsic activity, Antagonists, have affinity for the receptor. The binding of a drug to a receptor is determined by the following forces: 1. Hydrogen bonds 1. Ionic bonds 1. Van der Waals forces
Pharmacodynamics Factors Governing Drug-Receptor Interactions 1. Affinity (the ability of the drug to bind to the receptor). 2. Intrinsic activity = efficacy (the ability the drug to produce a detectable effect). Agonists have both affinity as well as intrinsic activity, Antagonists, have affinity for the receptor. The binding of a drug to a receptor is determined by the following forces: 1. Hydrogen bonds 1. Ionic bonds 1. Van der Waals forces
 Types of drugs Drugs can be classified as being either agonists or antagonists. Full Agonists: Compounds those are able to elicit a maximal response following receptor occupation and activation. Partial Agonists: Compounds that can activate receptors but are unable to elicit the maximal response of the receptor system. Antagonists (blockers): have the ability to bind to the receptor but do not initiate a change in cellular function. 1. Chemical Ant. : interact with the agonist away from the receptor site 2. Competitive Ant. : compete with agonists for the same recognition site of the receptors. Antagonism may be reversible or pseudo-irreversble 3. Non-competitive Ant. : bind to an allosteric site on the receptor different from the agonist recognition site. Antagonism may be reversible or irreversible 4. Functional Ant. : interfere with some processes subsequent to agonist activation of the receptor
Types of drugs Drugs can be classified as being either agonists or antagonists. Full Agonists: Compounds those are able to elicit a maximal response following receptor occupation and activation. Partial Agonists: Compounds that can activate receptors but are unable to elicit the maximal response of the receptor system. Antagonists (blockers): have the ability to bind to the receptor but do not initiate a change in cellular function. 1. Chemical Ant. : interact with the agonist away from the receptor site 2. Competitive Ant. : compete with agonists for the same recognition site of the receptors. Antagonism may be reversible or pseudo-irreversble 3. Non-competitive Ant. : bind to an allosteric site on the receptor different from the agonist recognition site. Antagonism may be reversible or irreversible 4. Functional Ant. : interfere with some processes subsequent to agonist activation of the receptor
 Adverse reactions of drugs These are harmful effects of a drug occurring at doses used for therapeutic, prophylactic or diagnostic purposes and which call for reduction of dose, drug withdrawal and/or immediate treatment. Adverse reactions may be type A (side effects and over dosage toxicity) or type B (hypersensitivity and idiosyncrasy). Adverse reactions may be related to the patient as age, sex, tendency to allergy, disease, personality and habits or to interactions between drugs. Adverse reactions include: 1. Side effects: Unavoidable part of the pharmacologic actions of the drug used for a specific indication at therapeutic doses as sedation with antihistaminics. 2. Over dosage toxicity: occur at high doses and their incidence increases as the dose is increased as liver damage from paracetamol overdosage, ototoxicity from aminoglycosides. 3. Allergic reactions: adverse reactions that are not dose-related, usually induced by prior contact with drugs that act as antigens. 4. Drug abuse: the use for non-therapeutic purpose of drugs that act on the CNS leading to dependence.
Adverse reactions of drugs These are harmful effects of a drug occurring at doses used for therapeutic, prophylactic or diagnostic purposes and which call for reduction of dose, drug withdrawal and/or immediate treatment. Adverse reactions may be type A (side effects and over dosage toxicity) or type B (hypersensitivity and idiosyncrasy). Adverse reactions may be related to the patient as age, sex, tendency to allergy, disease, personality and habits or to interactions between drugs. Adverse reactions include: 1. Side effects: Unavoidable part of the pharmacologic actions of the drug used for a specific indication at therapeutic doses as sedation with antihistaminics. 2. Over dosage toxicity: occur at high doses and their incidence increases as the dose is increased as liver damage from paracetamol overdosage, ototoxicity from aminoglycosides. 3. Allergic reactions: adverse reactions that are not dose-related, usually induced by prior contact with drugs that act as antigens. 4. Drug abuse: the use for non-therapeutic purpose of drugs that act on the CNS leading to dependence.
 Adverse reactions of drugs Idiosyncrasy: Inherent qualitatively abnormal reaction to a drug due to genetic abnormality as favism that occurs after Aspirin administration in glucose-6 phosphate dehydrogenase deficient person. Iatrogenic diseases (drug-induced diseases): A drug prescribed for a disease causes another disease as drug- induced asthma, peptic ulcer or parkinsonism Teratogenesis: Foetal abnormalities caused by some drugs when given early in pregnancy as anticancer drugs (abortion or foetal anomalies), tetracyclines (dental enamel hypoplasia). Drug allergy: A drug, metabolite or a non-drug element in the formulation activates the immune system in an undesirable way that appears as drug allergy. It is dose independent and occurs in minority of patients. Cross allergy occurs within a group of drugs. The chief target organs of drug allergy include skin, respiratory tract, GIT, blood and blood vessels.
Adverse reactions of drugs Idiosyncrasy: Inherent qualitatively abnormal reaction to a drug due to genetic abnormality as favism that occurs after Aspirin administration in glucose-6 phosphate dehydrogenase deficient person. Iatrogenic diseases (drug-induced diseases): A drug prescribed for a disease causes another disease as drug- induced asthma, peptic ulcer or parkinsonism Teratogenesis: Foetal abnormalities caused by some drugs when given early in pregnancy as anticancer drugs (abortion or foetal anomalies), tetracyclines (dental enamel hypoplasia). Drug allergy: A drug, metabolite or a non-drug element in the formulation activates the immune system in an undesirable way that appears as drug allergy. It is dose independent and occurs in minority of patients. Cross allergy occurs within a group of drugs. The chief target organs of drug allergy include skin, respiratory tract, GIT, blood and blood vessels.
 Adverse reactions of drugs Tolerance: Tolerance is an acquired resistance to the ordinary doses of drugs. It develops upon repeated administration and more drug is needed to produce the same effect. It disappears when the drug is stopped for sometime. Mechanisms of tolerance may be Increased metabolism due to enzyme induction, Development of antihormones (against insulin for example), Decreased sensitivity of receptors, Reduced efficacy at receptor sites (e. g. opiates), Down regulation of receptors (decreased number of receptors), occurs with B-agonists. Intolerance: A low threshold to the normal pharmacological action of a drug. . Cross tolerance: There is a decrease in response to one drug due to exposure to another drug. It is observed in treatment with antivirals, antibiotics, and many other medications. Tachyphylaxis (acute tolerance): It is a rapid form of tolerance in which we cannot get the same response by increasing the dose. Amphetamine and Ephedrine produce tachyphylaxis due to depletion of catecholamine stores and may be due to down regulation.
Adverse reactions of drugs Tolerance: Tolerance is an acquired resistance to the ordinary doses of drugs. It develops upon repeated administration and more drug is needed to produce the same effect. It disappears when the drug is stopped for sometime. Mechanisms of tolerance may be Increased metabolism due to enzyme induction, Development of antihormones (against insulin for example), Decreased sensitivity of receptors, Reduced efficacy at receptor sites (e. g. opiates), Down regulation of receptors (decreased number of receptors), occurs with B-agonists. Intolerance: A low threshold to the normal pharmacological action of a drug. . Cross tolerance: There is a decrease in response to one drug due to exposure to another drug. It is observed in treatment with antivirals, antibiotics, and many other medications. Tachyphylaxis (acute tolerance): It is a rapid form of tolerance in which we cannot get the same response by increasing the dose. Amphetamine and Ephedrine produce tachyphylaxis due to depletion of catecholamine stores and may be due to down regulation.
 Drug interactions Drug interactions: Pharmacologic responses which cannot be explained by the action of one drug but are due to multiple drugs acting concurrently. Drug interactions may result in antagonism or synergism. 1. Antagonism may be physiologic (as histamine/adrenaline), competitive (naloxone/morphine) or non-competitive (irreversible Ch. EIs). 2. Synergism may be in the form of summation (additive effect 1 + 1= 2) as alcohol and ether or potentiation (the combined effect is more powerful 1 + 1 > 2) as trimethoprim-sulphonamide combination. Drug interactions may be 1. pharmaceutical incompatibilities: occurring outside the body, 2. pharmacokinetic interactions: at sites of absorption, distribution, metabolism and excretion or 3. pharmacodynamic interactions: at sites of action. Examples of drug interactions are drug-food interactions. co-administering probenecid with penicillin.
Drug interactions Drug interactions: Pharmacologic responses which cannot be explained by the action of one drug but are due to multiple drugs acting concurrently. Drug interactions may result in antagonism or synergism. 1. Antagonism may be physiologic (as histamine/adrenaline), competitive (naloxone/morphine) or non-competitive (irreversible Ch. EIs). 2. Synergism may be in the form of summation (additive effect 1 + 1= 2) as alcohol and ether or potentiation (the combined effect is more powerful 1 + 1 > 2) as trimethoprim-sulphonamide combination. Drug interactions may be 1. pharmaceutical incompatibilities: occurring outside the body, 2. pharmacokinetic interactions: at sites of absorption, distribution, metabolism and excretion or 3. pharmacodynamic interactions: at sites of action. Examples of drug interactions are drug-food interactions. co-administering probenecid with penicillin.

 Types of Nervous System
Types of Nervous System
 Some anatomic and neurotransmitter features of peripheral nervous system
Some anatomic and neurotransmitter features of peripheral nervous system
 Efferent nerves of the peripheral nervous system
Efferent nerves of the peripheral nervous system
 The major components of the central and peripheral nervous systems and their functional relationships. Stimuli from the environment convey information to processing circuits within the brain and spinal cord, which in turn interpret their significance and send signals to peripheral effectors that move the body and adjust the workings of its internal organs.
The major components of the central and peripheral nervous systems and their functional relationships. Stimuli from the environment convey information to processing circuits within the brain and spinal cord, which in turn interpret their significance and send signals to peripheral effectors that move the body and adjust the workings of its internal organs.
 A. Drugs Acting on Motor Systems Mechanisms for influencing skeletal muscle tone. Inhibition of neuromuscular transmission and electromechanical coupling
A. Drugs Acting on Motor Systems Mechanisms for influencing skeletal muscle tone. Inhibition of neuromuscular transmission and electromechanical coupling
") A. Drugs Acting on Motor Systems: Muscle Relaxants 1. Non-depolarizing muscle relaxants (competitive antagonist) d-tubocurarine Semi-synthetic compound. Only I. V. Competitive antagonist towards Ach on nicotinic receptors. Onset: about 4 min. Duration: about 30 min. Antidote: acetylcholinesterase inhibitor. Adverse effects: bronchospasm, urticaria and hypotension. Pancuronium Synthetic compound. 5 -fold more potent than d-tubocurarine, with somewhat longer duration of action. Not likely to cause bronchospasm, urticaria and hypotension. Adverse effects: increased heart rate and blood pressure Newer compounds are vecuronium, pipecuronium, alcuronium, gallamine, mivacurium, and atracurium.
A. Drugs Acting on Motor Systems: Muscle Relaxants 1. Non-depolarizing muscle relaxants (competitive antagonist) d-tubocurarine Semi-synthetic compound. Only I. V. Competitive antagonist towards Ach on nicotinic receptors. Onset: about 4 min. Duration: about 30 min. Antidote: acetylcholinesterase inhibitor. Adverse effects: bronchospasm, urticaria and hypotension. Pancuronium Synthetic compound. 5 -fold more potent than d-tubocurarine, with somewhat longer duration of action. Not likely to cause bronchospasm, urticaria and hypotension. Adverse effects: increased heart rate and blood pressure Newer compounds are vecuronium, pipecuronium, alcuronium, gallamine, mivacurium, and atracurium.
 Succinylcholine") A. Drugs Acting on Motor Systems: Muscle Relaxants 2. Depolarizing muscle relaxants (agonists) Succinylcholine (suxamethonium) Double ACh molecule. Only I. V. Synthetic compound. Agonist at endplate nicotinic cholinoceptors, but it produces muscle relaxation due to the persistent depolarization of the endplate and adjoining membrane regions. Duration: about 10 min. Adverse effects: hyperkalemia (risk of cardiac arrhythmias). prolonged muscle relaxation and apnea in patients with a genetic deficiency in pseudocholinesterase. Used at the start of anesthesia to facilitate intubation of the patient. Antidote: no specific antidote.
A. Drugs Acting on Motor Systems: Muscle Relaxants 2. Depolarizing muscle relaxants (agonists) Succinylcholine (suxamethonium) Double ACh molecule. Only I. V. Synthetic compound. Agonist at endplate nicotinic cholinoceptors, but it produces muscle relaxation due to the persistent depolarization of the endplate and adjoining membrane regions. Duration: about 10 min. Adverse effects: hyperkalemia (risk of cardiac arrhythmias). prolonged muscle relaxation and apnea in patients with a genetic deficiency in pseudocholinesterase. Used at the start of anesthesia to facilitate intubation of the patient. Antidote: no specific antidote.
 B. Drugs Acting on the Parasympathetic Nervous System Responses to activation of the parasympathetic: Activation of ocular parasympathetic fibers (miosis and accommodation of near vision). ↑ Secretion of saliva and intestinal fluids (promotes digestion of foodstuffs; transport of intestinal contents). Allowing a decreased tidal volume (↑ bronchomotor tone) and ↓ cardiac activity. ↑ wall tension by detrusor activation with a concurrent relaxation of sphincter tonus (micturition).
B. Drugs Acting on the Parasympathetic Nervous System Responses to activation of the parasympathetic: Activation of ocular parasympathetic fibers (miosis and accommodation of near vision). ↑ Secretion of saliva and intestinal fluids (promotes digestion of foodstuffs; transport of intestinal contents). Allowing a decreased tidal volume (↑ bronchomotor tone) and ↓ cardiac activity. ↑ wall tension by detrusor activation with a concurrent relaxation of sphincter tonus (micturition).
 as a transmitter: (release,") B. Drugs Acting on the Parasympathetic Nervous System Acetyl-choline (ACh) as a transmitter: (release, effects, and degradation ) During activation of the nerve membrane, Ca 2+ is thought to enter the axoplasm and to activate protein kinases. As a result, vesicles discharge their contents into the synaptic gap. At the postsynaptic effector cell membrane, ACh reacts with M 1 receptors on nerve cells, e. g. , in ganglia. M 2 receptors mediate Ach effects on the heart. M 3 receptors mediate Ach effects on gut and bronchi and glandular epithelia. Released ACh is rapidly hydrolyzed and inactivated by a specific Achesterase, present on pre- and postjunctional membranes, or by a less specific serum cholinesterase, a soluble enzyme present in serum and interstitial fluid.
B. Drugs Acting on the Parasympathetic Nervous System Acetyl-choline (ACh) as a transmitter: (release, effects, and degradation ) During activation of the nerve membrane, Ca 2+ is thought to enter the axoplasm and to activate protein kinases. As a result, vesicles discharge their contents into the synaptic gap. At the postsynaptic effector cell membrane, ACh reacts with M 1 receptors on nerve cells, e. g. , in ganglia. M 2 receptors mediate Ach effects on the heart. M 3 receptors mediate Ach effects on gut and bronchi and glandular epithelia. Released ACh is rapidly hydrolyzed and inactivated by a specific Achesterase, present on pre- and postjunctional membranes, or by a less specific serum cholinesterase, a soluble enzyme present in serum and interstitial fluid.
 B. Drugs Acting on the Parasympathetic Nervous System 1. Parasympathomimetics ACh is too rapidly hydrolyzed and inactivated by ACh. E to be of any therapeutic use; however, its action can be mimicked by other substances, namely 1. Direct Parasympathomimetics as Carbachol, Pilocarpine and Arecoline (refreshing & mild stimulation betel chewing). 2. Indirect Parasympathomimetics as Esters of carbamic acid (carbamates such as physostigmine and neostigmine) and Phosphoric acid (organophosphates such as paraoxon and parathion. The rate-limiting step in ACh hydrolysis is deacetylation of the enzyme, which takes only milliseconds, thus permitting a high turnover rate and activity of ACh. E. Decarbaminoyl-ation following hydrolysis of carbamates takes hours to days, the enzyme remaining inhibited as long as it is carbaminoylated. Cleavage of the phosphate residue, i. e. dephosphoryl-ation, is practically impossible; enzyme inhibition is irreversible. Pilocarpine
B. Drugs Acting on the Parasympathetic Nervous System 1. Parasympathomimetics ACh is too rapidly hydrolyzed and inactivated by ACh. E to be of any therapeutic use; however, its action can be mimicked by other substances, namely 1. Direct Parasympathomimetics as Carbachol, Pilocarpine and Arecoline (refreshing & mild stimulation betel chewing). 2. Indirect Parasympathomimetics as Esters of carbamic acid (carbamates such as physostigmine and neostigmine) and Phosphoric acid (organophosphates such as paraoxon and parathion. The rate-limiting step in ACh hydrolysis is deacetylation of the enzyme, which takes only milliseconds, thus permitting a high turnover rate and activity of ACh. E. Decarbaminoyl-ation following hydrolysis of carbamates takes hours to days, the enzyme remaining inhibited as long as it is carbaminoylated. Cleavage of the phosphate residue, i. e. dephosphoryl-ation, is practically impossible; enzyme inhibition is irreversible. Pilocarpine
 B. Drugs Acting on the Parasympathetic Nervous System 1. Parasympatho-mimetics Uses of parasympathomimetics In postoperative atonia of the bowel or bladder (neostigmine). In myasthenia gravis to overcome the relative ACh-deficiency at the motor endplate In de-curarization before discontinuation of anesthesia to reverse the neuromuscular blockade caused by non-depolarizing muscle relaxants. As antidote in poisoning with parasympatholytic drugs because it has access to ACh. E in the brain (physostigmine). In the treatment of glaucoma (neostigmine, pyridostigmine, physostigmine pilocarpine paraoxon and ecothiopate): however, their long-term use leads to cataract formation. Insecticides (parathion). Although they possess high acute toxicity in humans, they are more rapidly degraded than is the insecticide DDT following their emission into the environment. Tacrine is not an ester and interferes only with the choline-binding site of ACh. E. It is effective in alleviating symptoms of dementia in some subtypes of Alzheimer’s disease.
B. Drugs Acting on the Parasympathetic Nervous System 1. Parasympatho-mimetics Uses of parasympathomimetics In postoperative atonia of the bowel or bladder (neostigmine). In myasthenia gravis to overcome the relative ACh-deficiency at the motor endplate In de-curarization before discontinuation of anesthesia to reverse the neuromuscular blockade caused by non-depolarizing muscle relaxants. As antidote in poisoning with parasympatholytic drugs because it has access to ACh. E in the brain (physostigmine). In the treatment of glaucoma (neostigmine, pyridostigmine, physostigmine pilocarpine paraoxon and ecothiopate): however, their long-term use leads to cataract formation. Insecticides (parathion). Although they possess high acute toxicity in humans, they are more rapidly degraded than is the insecticide DDT following their emission into the environment. Tacrine is not an ester and interferes only with the choline-binding site of ACh. E. It is effective in alleviating symptoms of dementia in some subtypes of Alzheimer’s disease.
 B. Drugs Acting on the Parasympathetic Nervous System 2. Parasympatho-lytics Effects of parasympathetic stimulation (blue arrow) and blockade (red) Parasympathomimetics are substances acting agonistically at the M cholinoceptor (blue arrows). Parasympatholytics are substances acting antagonistically at the M cholinoceptor (shown in red in the panels).
B. Drugs Acting on the Parasympathetic Nervous System 2. Parasympatho-lytics Effects of parasympathetic stimulation (blue arrow) and blockade (red) Parasympathomimetics are substances acting agonistically at the M cholinoceptor (blue arrows). Parasympatholytics are substances acting antagonistically at the M cholinoceptor (shown in red in the panels).
) Uses of") B. Drugs Acting on the Parasympathetic Nervous System 2. Parasympatholytics ((Atropine-like drugs)) Uses of parasympatholytics: Atropine; is used to prevent cardiac arrest and as a preanesthetic medication to prevents a possible hypersecretion of bronchial mucus, which cannot be expectorated by coughing during anesthesia. Homatropine; is used as mydriatics (for diagnostic use). Benzatropine; is used in treatment of Parkinson’s disease. Pirenzepine; is used in treatment of gastric and duodenal ulcers. Ipratropium; is used in treatment of bronchial asthma, bradycardia and heart block. N-butylscopolamine; is used in treatment of biliary & renal colic. Scopolamine; is used in treatment of motion sickness. Contraindications for parasympatholytics: Glaucoma and Prostatic hypertrophy with impaired micturition. Atropine poisoning: Peripheral (tachycardia; dry mouth; hyperthermia, flashing and constipation) and Central ( restlessness, agitation, psychic disturbances and hallucinations) effects. Treatment of Atropine poisoning, general measures (gastric lavage, cooling with ice water) or therapy with indirect parasympathomimetic as physostigmine.
B. Drugs Acting on the Parasympathetic Nervous System 2. Parasympatholytics ((Atropine-like drugs)) Uses of parasympatholytics: Atropine; is used to prevent cardiac arrest and as a preanesthetic medication to prevents a possible hypersecretion of bronchial mucus, which cannot be expectorated by coughing during anesthesia. Homatropine; is used as mydriatics (for diagnostic use). Benzatropine; is used in treatment of Parkinson’s disease. Pirenzepine; is used in treatment of gastric and duodenal ulcers. Ipratropium; is used in treatment of bronchial asthma, bradycardia and heart block. N-butylscopolamine; is used in treatment of biliary & renal colic. Scopolamine; is used in treatment of motion sickness. Contraindications for parasympatholytics: Glaucoma and Prostatic hypertrophy with impaired micturition. Atropine poisoning: Peripheral (tachycardia; dry mouth; hyperthermia, flashing and constipation) and Central ( restlessness, agitation, psychic disturbances and hallucinations) effects. Treatment of Atropine poisoning, general measures (gastric lavage, cooling with ice water) or therapy with indirect parasympathomimetic as physostigmine.
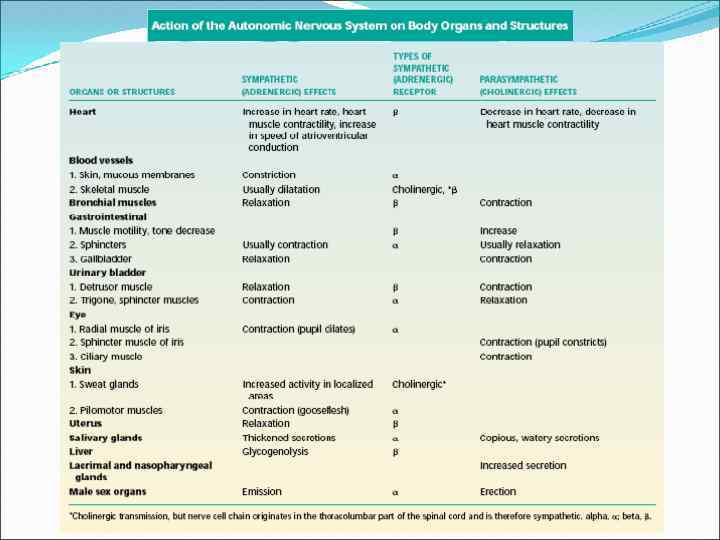
 C. Drugs Acting on the Sympathetic Nervous System Responses to sympathetic activation CNS Eye Saliva Bronchi Sweat glands Heart Liver Intestinal tract Bladder Skeletal muscle
C. Drugs Acting on the Sympathetic Nervous System Responses to sympathetic activation CNS Eye Saliva Bronchi Sweat glands Heart Liver Intestinal tract Bladder Skeletal muscle
 C. Drugs Acting on the Sympathetic Nervous System Sympathetic Transmitter nor-epinephrine (NE also called nor-adrenaline) Synthesis of nor-epinephrine Releases of nor-epinephrine Fate of nor-epinephrine 1. Neuronal re-uptake 2. Inactivated by MAO 3. Inactivated by COMT
C. Drugs Acting on the Sympathetic Nervous System Sympathetic Transmitter nor-epinephrine (NE also called nor-adrenaline) Synthesis of nor-epinephrine Releases of nor-epinephrine Fate of nor-epinephrine 1. Neuronal re-uptake 2. Inactivated by MAO 3. Inactivated by COMT
 Receptors") C. Drugs Acting on the Sympathetic Nervous System 1. Sympathomimetics Adrenoceptors (adrenergic receptors) Receptors Organs α 1 Smooth muscle (blood vessels, lung, intestine, bladder, , , , α 2 Pre-synaptic adrenergic nerve terminals, platelets, lipocytes, smooth muscle β 1 Heart, lipocytes, brain β 2 Smooth muscle and cardiac muscle Sympathomimetics (i. e. , adrenoceptor agonists) Direct-acting sympathomimetics Epinephrine (Adrenaline) Norepinephrine (Noradrenaline) Isoproterenol Phenylephrine Dobutamine Salbutamol Indirect-acting sympathomimetics Cocaine inhibit NE re-uptake Ephedrine facilitate NE releases Amphetamine All the above + slow breakdown by MAO Receptors α 1, α 2, β 1, β 2 α 1, α 2 > β 1, β 2 β 1 and β 2 α 1 β 2 α and β
C. Drugs Acting on the Sympathetic Nervous System 1. Sympathomimetics Adrenoceptors (adrenergic receptors) Receptors Organs α 1 Smooth muscle (blood vessels, lung, intestine, bladder, , , , α 2 Pre-synaptic adrenergic nerve terminals, platelets, lipocytes, smooth muscle β 1 Heart, lipocytes, brain β 2 Smooth muscle and cardiac muscle Sympathomimetics (i. e. , adrenoceptor agonists) Direct-acting sympathomimetics Epinephrine (Adrenaline) Norepinephrine (Noradrenaline) Isoproterenol Phenylephrine Dobutamine Salbutamol Indirect-acting sympathomimetics Cocaine inhibit NE re-uptake Ephedrine facilitate NE releases Amphetamine All the above + slow breakdown by MAO Receptors α 1, α 2, β 1, β 2 α 1, α 2 > β 1, β 2 β 1 and β 2 α 1 β 2 α and β
 C. Drugs Acting on the Sympathetic Nervous System 1. Sympathomimetics Clinical uses of adrenoceptor agonists 1. Cardiovascular system cardiac arrest as adrenaline cardiogenic shock as dobutamine heart block as isoprenaline, which can be used temporarily while electrical pacing is being arranged. 2. Anaphylactic shock (acute hypersensitivity): adrenaline is the first-line treatment 3. Respiratory system (asthma): selective β 2 -receptor agonists (salbutamol, formoterol) 4. Nasal decongestion: drops containing phenylephrine, oxymetazoline or ephedrine reduces mucosal blood flow and, hence, capillary pressure. Fluid exuded into the interstitial space is drained through the veins, thus shrinking the nasal mucosa. Due to the reduced supply of fluid, secretion of nasal mucus decreases. 5. Miscellaneous indications Adrenaline can be used to prolong local anaesthetic action by delaying the removal of local anesthetic. Inhibition of premature labour (salbutamol) α 2 -agonists as clonidine used in hypertension, menopausal flushing, lowering intraocular pressure and migraine prophylaxis.
C. Drugs Acting on the Sympathetic Nervous System 1. Sympathomimetics Clinical uses of adrenoceptor agonists 1. Cardiovascular system cardiac arrest as adrenaline cardiogenic shock as dobutamine heart block as isoprenaline, which can be used temporarily while electrical pacing is being arranged. 2. Anaphylactic shock (acute hypersensitivity): adrenaline is the first-line treatment 3. Respiratory system (asthma): selective β 2 -receptor agonists (salbutamol, formoterol) 4. Nasal decongestion: drops containing phenylephrine, oxymetazoline or ephedrine reduces mucosal blood flow and, hence, capillary pressure. Fluid exuded into the interstitial space is drained through the veins, thus shrinking the nasal mucosa. Due to the reduced supply of fluid, secretion of nasal mucus decreases. 5. Miscellaneous indications Adrenaline can be used to prolong local anaesthetic action by delaying the removal of local anesthetic. Inhibition of premature labour (salbutamol) α 2 -agonists as clonidine used in hypertension, menopausal flushing, lowering intraocular pressure and migraine prophylaxis.
 1. α-Sympatholytics") C. Drugs Acting on the Sympathetic Nervous System 2. Sympatholytics (Sympathatic blockers) 1. α-Sympatholytics (α-blockers) non-selective (blocks both post-synaptic and pre-synaptic α-adrenoceptors) as phentolamine. selective α 1 -blockers as prazosin and terazosin. α-blockers are used in treatment of hypertension and in benign hyperplasia of the prostate. side effects of α-blockers are postural hypotension. 2. β-Sympatholytics (β-Blockers) non-selective as propranolol. selective β 1 -receptors as metoprolol, acebutolol, bisoprolol. β-blockers are used in treatment of angina pectoris, tachycardia, hypertension, glaucoma. side effects of β-blockers are congestive heart failure, bradycardia, bronchial asthma, hypoglycemia in diabetes mellitus and sedation. 3. α and β blockers as Carvendilol used in treatment of congestive heart failure and hypertension. Labetalol used in treatment of hypertension. 4. Centrally acting anti-adrenergics drugs They are capable of lowering transmitter output from sympathetic neurons. Their action is hypotensive however, being poorly tolerated, they enjoy only limited therapeutic use. Examples are Clonidine, Methyldopa, Reserpine and Guanethidine.
C. Drugs Acting on the Sympathetic Nervous System 2. Sympatholytics (Sympathatic blockers) 1. α-Sympatholytics (α-blockers) non-selective (blocks both post-synaptic and pre-synaptic α-adrenoceptors) as phentolamine. selective α 1 -blockers as prazosin and terazosin. α-blockers are used in treatment of hypertension and in benign hyperplasia of the prostate. side effects of α-blockers are postural hypotension. 2. β-Sympatholytics (β-Blockers) non-selective as propranolol. selective β 1 -receptors as metoprolol, acebutolol, bisoprolol. β-blockers are used in treatment of angina pectoris, tachycardia, hypertension, glaucoma. side effects of β-blockers are congestive heart failure, bradycardia, bronchial asthma, hypoglycemia in diabetes mellitus and sedation. 3. α and β blockers as Carvendilol used in treatment of congestive heart failure and hypertension. Labetalol used in treatment of hypertension. 4. Centrally acting anti-adrenergics drugs They are capable of lowering transmitter output from sympathetic neurons. Their action is hypotensive however, being poorly tolerated, they enjoy only limited therapeutic use. Examples are Clonidine, Methyldopa, Reserpine and Guanethidine.
 C. Drugs Acting on the Sympathetic Nervous System 2. Sympatholytics Clonidine. Activates post-synaptic and pre-synaptic α 2 -receptors and leads to a decreased release of nor-epinephrine (NE). Side effects. Dry mouth; rebound hypertension after abrupt cessation of clonidine therapy. Methyldopa. It converts in the brain to α-methyl-dopamine, and then to α-methyl-NE thus competes for a portion of the available enzymatic activity (inhibition of Dopa-decarboxylase), so that the rate of conversion of L-dopa to NE (via dopamine) is decreased. The false transmitter α-methyl-NE can be stored; however, unlike the endogenous mediator, it has a higher affinity for α 2 - than for α 1 -receptors and therefore produces effects similar to those of clonidine. The same events take place in peripheral adrenergic neurons. Adverse effects. Fatigue, orthostatic hypotension, extrapyramidal Parkinson-like symptoms, hepatic damage, immune-hemolytic anemia.
C. Drugs Acting on the Sympathetic Nervous System 2. Sympatholytics Clonidine. Activates post-synaptic and pre-synaptic α 2 -receptors and leads to a decreased release of nor-epinephrine (NE). Side effects. Dry mouth; rebound hypertension after abrupt cessation of clonidine therapy. Methyldopa. It converts in the brain to α-methyl-dopamine, and then to α-methyl-NE thus competes for a portion of the available enzymatic activity (inhibition of Dopa-decarboxylase), so that the rate of conversion of L-dopa to NE (via dopamine) is decreased. The false transmitter α-methyl-NE can be stored; however, unlike the endogenous mediator, it has a higher affinity for α 2 - than for α 1 -receptors and therefore produces effects similar to those of clonidine. The same events take place in peripheral adrenergic neurons. Adverse effects. Fatigue, orthostatic hypotension, extrapyramidal Parkinson-like symptoms, hepatic damage, immune-hemolytic anemia.
 C. Drugs Acting on the Sympathetic Nervous System 2. Sympatholytics Reserpine. It abolishes the vesicular storage of biogenic amines (NE, dopamine, serotonin = 5 -HT). To a lesser degree, release of epinephrine from the adrenal medulla is also impaired. Adverse effects. Disorders of extrapyramidal motor function with development of pseudo-Parkinsonism, sedation, depression, stuffy nose, impaired libido, and impotence; increased appetite. These adverse effects have rendered the drug practically obsolete.
C. Drugs Acting on the Sympathetic Nervous System 2. Sympatholytics Reserpine. It abolishes the vesicular storage of biogenic amines (NE, dopamine, serotonin = 5 -HT). To a lesser degree, release of epinephrine from the adrenal medulla is also impaired. Adverse effects. Disorders of extrapyramidal motor function with development of pseudo-Parkinsonism, sedation, depression, stuffy nose, impaired libido, and impotence; increased appetite. These adverse effects have rendered the drug practically obsolete.
 C. Drugs Acting on the Sympathetic Nervous System 2. Sympatholytics Guanethidine. It has high affinity for the axolemmal and vesicular amine transporters. It is stored instead of NE, but is unable to mimic the functions of the latter. In addition, it stabilizes the axonal membrane, thereby impeding the propagation of impulses into the sympathetic nerve terminals. Storage and release of epinephrine from the adrenal medulla are not affected, owing to the absence of a re-uptake process. The drug does not cross the bloodbrain barrier. Adverse effects. Cardiovascular crises are a possible risk: emotional stress of the patient may cause sympatho-adrenal activation with epinephrine release. The resulting rise in blood pressure can be all the more marked because persistent depression of sympathetic nerve activity induces supersensitivity of effector organs to circulating catecholamines.
C. Drugs Acting on the Sympathetic Nervous System 2. Sympatholytics Guanethidine. It has high affinity for the axolemmal and vesicular amine transporters. It is stored instead of NE, but is unable to mimic the functions of the latter. In addition, it stabilizes the axonal membrane, thereby impeding the propagation of impulses into the sympathetic nerve terminals. Storage and release of epinephrine from the adrenal medulla are not affected, owing to the absence of a re-uptake process. The drug does not cross the bloodbrain barrier. Adverse effects. Cardiovascular crises are a possible risk: emotional stress of the patient may cause sympatho-adrenal activation with epinephrine release. The resulting rise in blood pressure can be all the more marked because persistent depression of sympathetic nerve activity induces supersensitivity of effector organs to circulating catecholamines.
 nerve (causes skeletal muscle contraction) The neurotransmitter is acetyl-choline (ACh). ACh is") Somatic (motor) nerve (causes skeletal muscle contraction) The neurotransmitter is acetyl-choline (ACh). ACh is hydrolyzed by acetyl-cholin-esterase. Receptor is Nicotinic (N). Convulsant as tetanus toxin, strychnine. Centrally acting muscle relaxants as benzo-diazepines, baclofen, clonidine. Non-depolarizing muscle relaxants as curare, d-tubo-curarine, pan-curonium. Depolarizing muscle relaxants as succinyl-choline. Parasympathetic nervous system (like when you eat) The neurotransmitter is acetyl-choline (ACh). Receptors are Muscarinic (M) and Nicotinic (N) receptors. ACh is hydrolyzed by acetyl-cholin-esterase. Parasympatho-mimetics may be direct as carbachol, pilocarpine, arecoline or indirect as physostigmine, neostigmine, paraoxon, parathion. Parasympatho-lytics as atropine, hom-atropine, benz-atropine, pirenzepine, ipr-atropium, N-butylscopolamine, scopolamine. Sympathetic nervous system (like when you play football) The neurotransmitter is nor-epinephrine (NE). Receptors are α and β receptors. NE is decreased at the receptors by neuronal re-uptake, MAO or COMT. Sympatho-mimetics may be direct as epinephrine (adrenaline) nor-epinephrine (nor-adrenaline), isoproterenol, phenyl-ephrine, dobutamine, salbutamol or indirect as cocaine, ephedrine, amphetamine. Sympatho-lytics as phentol-amine (post-synaptic and pre-synaptic α receptors blocker), prazosin (α 1), propranolol (β 1 and β 2), metoprolol, acebutolol, bisoprolol (β 1 > β 2), carvendilol and labetalol (α and β), clonidine (pre-synaptic α 2 -agonist), methyldopa (act as a false transmitter α-methyl-NE), reserpine (abolishes the vesicular storage of NE), guanethidine (stored instead of NE and stabilizes the axonal membrane).
Somatic (motor) nerve (causes skeletal muscle contraction) The neurotransmitter is acetyl-choline (ACh). ACh is hydrolyzed by acetyl-cholin-esterase. Receptor is Nicotinic (N). Convulsant as tetanus toxin, strychnine. Centrally acting muscle relaxants as benzo-diazepines, baclofen, clonidine. Non-depolarizing muscle relaxants as curare, d-tubo-curarine, pan-curonium. Depolarizing muscle relaxants as succinyl-choline. Parasympathetic nervous system (like when you eat) The neurotransmitter is acetyl-choline (ACh). Receptors are Muscarinic (M) and Nicotinic (N) receptors. ACh is hydrolyzed by acetyl-cholin-esterase. Parasympatho-mimetics may be direct as carbachol, pilocarpine, arecoline or indirect as physostigmine, neostigmine, paraoxon, parathion. Parasympatho-lytics as atropine, hom-atropine, benz-atropine, pirenzepine, ipr-atropium, N-butylscopolamine, scopolamine. Sympathetic nervous system (like when you play football) The neurotransmitter is nor-epinephrine (NE). Receptors are α and β receptors. NE is decreased at the receptors by neuronal re-uptake, MAO or COMT. Sympatho-mimetics may be direct as epinephrine (adrenaline) nor-epinephrine (nor-adrenaline), isoproterenol, phenyl-ephrine, dobutamine, salbutamol or indirect as cocaine, ephedrine, amphetamine. Sympatho-lytics as phentol-amine (post-synaptic and pre-synaptic α receptors blocker), prazosin (α 1), propranolol (β 1 and β 2), metoprolol, acebutolol, bisoprolol (β 1 > β 2), carvendilol and labetalol (α and β), clonidine (pre-synaptic α 2 -agonist), methyldopa (act as a false transmitter α-methyl-NE), reserpine (abolishes the vesicular storage of NE), guanethidine (stored instead of NE and stabilizes the axonal membrane).
 Sources Report items Dosage forms (solutions, suspensions, emulsions, tablets, coated . 1. 2 Pharmacokinetics (absorption, distribution, metabolism, . 3 Pharmacodynamics (mechanisms of action and effects on body . 4 Adverse reactions Contraindications Drug interactions . 5. 6. 7 (synthetic, semi-synthetic, natural). tablets, capsules, injections, aerosols, suppositories, powders, ointments, pastes). elimination). Binding to plasma proteins, Renal clearance, Elimination half life. organs).
Sources Report items Dosage forms (solutions, suspensions, emulsions, tablets, coated . 1. 2 Pharmacokinetics (absorption, distribution, metabolism, . 3 Pharmacodynamics (mechanisms of action and effects on body . 4 Adverse reactions Contraindications Drug interactions . 5. 6. 7 (synthetic, semi-synthetic, natural). tablets, capsules, injections, aerosols, suppositories, powders, ointments, pastes). elimination). Binding to plasma proteins, Renal clearance, Elimination half life. organs).