
Презентация alport end















- Размер: 515.5 Кб
- Количество слайдов: 17
Описание презентации Презентация alport end по слайдам
25. 12. 2010 Синдром Альпорта Карпов Дмитрий Оленчук Владислав Огороднищук Максим Щербакова Ольга
25. 12. 2010 Синдром Альпорта. Историческаясправка: Первое упоминаниепатологии, известнойкаксиндром. Альпорта, принадлежит L. Guthrie, которыйв 1902 г. описалсемьюсгематуриейвнесколькихпоколениях. A. Hurst в 1915 г. вэтойжесемьенаблюдалразвитиеуремии. В 1927 г. A. Alport, описывая глухотуунесколькихродственниковсгематурией, отметил, чтоу мужчин уремияразвиваласьраньше, чемуженщин. В 1972 г. выявленонеравномерноерасширениеирасслоениеплотнойпластинки гломерулярных базальныхмембранприсиндроме. Альпорта, выраженность которых коррелироваласвозрастомиполом, определяяпрогрессирование болезни. В 70 егоды. М. С. Игнатовойи. В. В. Фокеевойнаосновенаблюденияза 200 детьми высказывалась гипотезаоважнейшейроливразвитиинаследственногонефрита патологии соединительнойткани, вкачествекритериясостояниягломерулярных базальных мембранисследоваласьэкскрециягидроксилизингликозидов.
25. 12. 2010 Синдром Альпорта. Генетика: Гены : Col. IVAIII Col. IVAIV Col. IVAVI Расположение хромосомы: Col. IVAIII, AIV локализуетсяна 2 хромосомечеловекавпозицииq 3537 Col. IVAV локализуетсяв. Ххромосомевпозицииq 2223 Наследственность : C индром. Альпортанаследуетсяпосцепленномус. Ххромосомой доминантному илирецессивномутипу. IIIтип, илиаутосомнодоминантному, или аутосомнорецессивномутипу. Iи. IIтип Тип клеток, местоэкспрессии: Базальная гломерулярнаямембрана. Этобесклеточныйматрикстолщинойв 300500 нм, который представляетсобойструктурнуюопорудлякапиллярнойстенки. Ееглавными компонентами являютсяколлаген. IVтипа, протеогликаны, ламинининидоген. Коллаген плода свозрастомзаменяетсяколлагеномвзрослых. Вслучаяхмутациипроисходит искажение структурыбазальноймембраныгломерулубольныхсиндромом. Альпорта, что ведет кпоявлениюгематурии, какпризнакапочечнойпатологии, агематурияявляется причиной легкойпротеинурии.
25. 12. 2010 Синдром Альпорта. COL 4 A 5 COL
25. 12. 2010 Синдром Альпорта. Генетика: Число итипмутаций: Коллаген IVтипасостоитизтрехдоменов, плотноскрученныхдругсдругомвнормальном состоянии. Примутациигена(чаще. COL 4 A 5)наблюдаютсяаномалиискручивания коллагена IVтипа. Еслинаблюдаетсяделеция. COL 4 A 5, чтобылоотмеченонамив. ДНК 5 из 16 обследованныхсемей, происходитвыпадениебольшойчастиэкзоновгена. Вэтих случаях нарушаетсяскручиваниедоменов. Заболеваниеимееттяжелоепрогрессирующее течение сразвитиемэкстраренальныхпроявленийиформированием. ХПН. Также наблюдается резкаядеформациягломерулярных. БМ. Приэлектронноймикроскопии(ЭМ) почечного биоптатаотмечаютсявыраженныедистрофическиеизменения. БМсучастками просветления ископлениемтонкогранулярноговещества. Этоприводиткпоявлению протеинурии споследующимиизменениямивсистемеангиотензина. IIи трансформирующего фактораростабета(ТФР ). Экспериментальныеданныеβ подтверждают значимость. ТФР впрогрессированиисиндрома. Альпорта]. Втехслучаях, β когда отмечаетсятолькооднонуклеотиднаязаменавгене, т. е. происходитточечная мутация , нарушенияскручиванияколлагенавыявляютсялишьвотдельныхучастках, БМ остается тонкойиклиническиубольногоимеетместоизолированнаягематурия. Именно подобные случаи. САи. БТБМпредставляютбольшиесложностиприпроведении дифференциального диагноза.
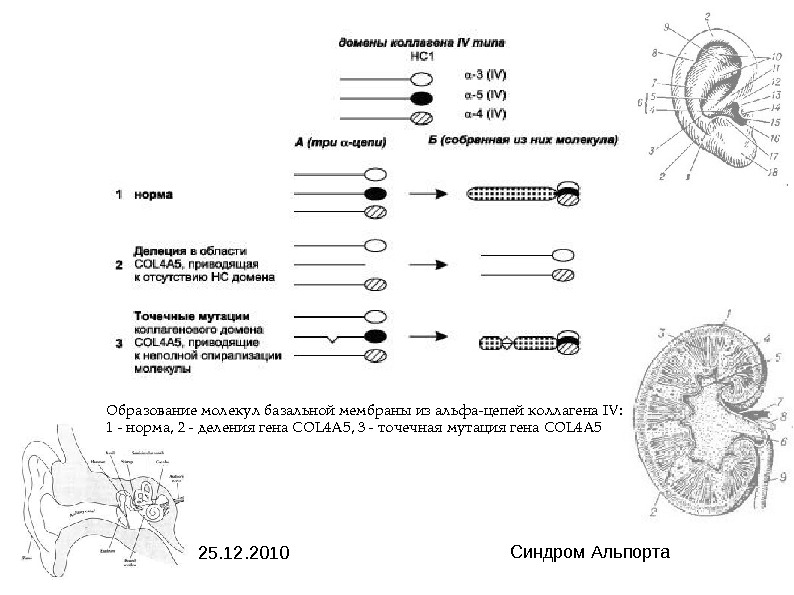
25. 12. 2010 Синдром Альпорта. Образованиемолекулбазальноймембраныизальфацепейколлагена. IV: 1 норма, 2 делениягена. COL 4 A 5, 3 точечнаямутациягена. COL
25. 12. 2010 Синдром Альпорта. Белок: Состав : Коллаген 4 типа Состав Col. IVAIII 1260 аминокислотныхостатков Col. IVAIV 1690 аминокислотныхостатков, включая 38 аминокислот сигнального пептида, отщепляющегосявпроцессесозревания Расположение : Col. IV находитсявбазальныхмембранах. Базальнаямембранаимеет толщину около 1 мкмисостоитиздвухпластинок: светлой(laminalucida) и темной(laminadensa). Основнымикомпонентамибазальныхмембран являются коллаген. IVтипа, ламинин, гепарансульфатсодержащие протеогликаны (ГСПГ). При этомнерастворимостьимеханическуюстабильностьбазальных мембран обеспечиваютмолекулыколлагена. IVтипа, которые организуются вопорнуюсеть. Этаэластичнаятрёхмернаясетьобразует структурный остов, ккоторомуприкрепляютсядругиекомпоненты базальных мембран. В почечныхклубочкахбазальнаямембранаслужитполупроницаемым фильтром , препятствующимпереходумакромолекулизплазмыв первичную мочу.
25. 12. 2010 Синдром Альпорта. Белок: Структурные особенности: Молекула коллагенапредставляетсобойправозакрученнуюспиральизтрёх цепей. α Один витокспирали цеписодержиттриаминокислотныхостатка. α Молекулярная массаколлагенаоколо 300 к. Да, длина 300 нм, толщина 1, 5 нм. цеписостоитизтриадаминокислот. Втриадахтретьяаминокислотавсегдаα глицин , вторая—пролинилилизин, первая—любаядругаяаминокислота, кроме трёхперечисленных. Первичная структураколлагенахарактеризуетсяпоследовательным расположением аминокислотныхостатковиихколичествомвегополипептидных цепях. Аминокислотымогутотноситьсякалифатическим, карбоциклическими гетероциклическим. Взависимостиотстроениябоковойцепиаминокислотные остатки подразделяютнатипы, составкоторых, %отобщегочисла аминокислотных остатков, данниже. Без боковойцепи(гликоколь)33, 34 С гидрофильнойбоковойцепью: кислотного характера(аспарагиноваяиглютаминоваяаминокислоты)12, 38 основного характера(лизин, оксилизин, аргинин, гистидин)8, 96 Серосодержащие (метионин)0, 70 Содержащие гидроксил, заисключениемоксилизина ( оксипролин, тирозин, серии, треонин)13, 54 Не содержащиеазотикислородвбоковойцепи ( аланин, лейцин, изолейцин, валин, фенилаланин, пролин)31, 48 С иминогруппой(пролиниоксипролин)21, 40 Элементы вторичнойструктурыколлагена–спиральныеполипептидыбелка, так называемые цепи, –являютсяосновнойединицейтретичнойструктуры–α тропоколлагеновой частицы, состоящейизтрехполипептидныхцепейсобщей осью.
25. 12. 2010 Синдром Альпорта
25. 12. 2010 Синдром Альпорта. Белок: Функциональность : Белок (молекулыколлагена. IVтипа)обеспечиваетнерастворимостьи механическую стабильностьбазальныхмембран, которыеорганизуютсяв опорную сеть. Этаэластичнаятрёхмернаясетьобразуетструктурныйостов, к которому прикрепляютсядругиекомпонентыбазальныхмембран. В почечныхклубочкахбазальнаямембранаслужитполупроницаемым фильтром , препятствующимпереходумакромолекулизплазмывпервичную мочу. В какихорганизмахприсутствует: Колаген четвертоготипасодержитсяпреимущественновэукариотах.
25. 12. 2010 Синдром Альпорта. Признакиболезниворганизме: Основные симптомы: Клиническая картинасиндрома. Альпорта, регулярноповторяющаясявсемье, обычно соответствует какомулибофенотипу, хотявыраженностьсимптомовможетменятьсяот человека кчеловекуивзависимостиотвозрастаипола. Большинствосемейсэтой патологией хорошовписываютсявследующуюклассификацию: • Доминантный юношескийнефритстугоухостью. • Х сцепленныйнефритстугоухостьюувзрослых. • Х сцепленныйнефритбезэкстраренальныхпроявлений. • Аутосомно доминантныйнефритстугоухостьюитромбоцитопатией, соответствующий по Mc. Kusickкатегории. N 15365(синдром. Эпштейна). • Аутосомно доминантныйнефритюношескоготипастугоухостью. Юношеским вариантомсиндрома. Альпортасчитаютсятеслучаи, когдахроническая почечная недостаточностьразвиваетсяраньше 31 года.
25. 12. 2010 Синдром Альпорта. Признакиболезниворганизме: Связь сгенетическимизаболеваниями: При болезни. Шарко. Мари. Тутасемейноесочетаниенефропатии, тугоухости, фокально сегментарногогломерулосклерозаирасслоенияплотнойпластинки гломерулярных базальныхмембрансопровождаетсямышечнойатрофией. Нефропатияи тугоухость присиндроме. Branchio. Oto. Renalсочетаетсясрудиментарнымиостатками жаберных щелей. Длясиндрома. Макла. Уэльсахарактерныаутосомнодоминантное наследование , повышение. СОЭ, частоеразвитиехроническойпочечнойнедостаточности, озноб иуртикарнаясыпь(вдебюте), тугоухость, глаукомаинефротическийсиндром ( впоследствии). Присиндроме. Альстремапигментнаядегенерациясетчатки, нейросенсорная тугоухостьинефропатиясочетаютсяссахарнымдиабетомиожирением. Синдром Sebastianтруднодифференцируетсяот. Vтипасиндрома. Альпортавсвязис общей гематологическойкартиной. В сочетаниистугоухостьюописаныинтерстициальныйнефритаутосомнодоминантного генеза сразвитиемхроническойпочечнойнедостаточностивзреломвозрасте, почечный тубулярный ацидозисемейныеслучаи. Ig. Aнефропатии. Средисемейныхслучаев гломерулонефрита чащевстречаютсянаблюденияаутосомнодоминантногонаследования этой патологии. Гематуриейсопровождаетсяиврожденныйдефицит3 йфракции комплемента. Хсцепленноенаследованиепрогрессирующейтугоухостичасто мимикрирует синдром. Альпортавсвязисаллельностьюихгенов
25. 12. 2010 Синдром Альпорта. Признакиболезниворганизме: Типы пораженныхклеток: Нейроны , волосяныеклетки Необычные особенностиболезни: • Поражение нервов(полиневропатия), • Миастения , • Потеря памятииинтеллекта, • Тромбоцитопения. Модель животногодлязаболевания: Белые крысыбеспороднойлинии.
25. 12. 2010 Синдром Альпорта. Клеточнаяимолекулярнаябиология: Поражение органелл: Поражаются базальныемембраны. Базальная мембранаэтоплотноебесклеточноеобразование, накотором располагаются клеткиэпителияилиэндотелия. Всоставбазальноймембраны входят гликопротеиды, гликозаминогликаныиколлаген. Базальнаямембрана выполняет опорнуюфункцию, поддерживаяформуоргановисосудов. Патология тканейиклиническиеособенностиприсиндроме. Альпортеэто результат , экспрессииколлагена 3, 4, 5, и, возможно, 6(IV)цепейвα α базальной мембране. Этицепи, какправило, отсутствуетилинедостаточно выраженные вбазальныхмембранахлицссиндромом. Альпорте, такчтосети которые ониобразуютотсутствуютили, еслиониприсутствуют, дефективныпо структуре ифункции.
25. 12. 2010 Синдром Альпорта. Клеточнаяимолекулярнаябиология: Нормальные функции: В нормальноразвивающейсяпочке, изначальноколлаген 1(IV)иколлаген 2(IV)α α цепи преобладаютвгломерулярнойбазальноймембраненезрелыхпочечных клубочках. Формированиекапиллярныхпетельвтечениесозреванияклубочков связано споявлениемколлагена 3, 4, и 5(IV)цепейвгломерулярнойα α α базальной мембраны. Втовремякаксозреваниепрогрессирует, 3, 4, и 5(IV)α α α цепи становятсяпреобладающимтипомколлагена. IVвцепяхгломерулярной базальной мембраны. Этотпроцессбылназванкак»изотипныепереключения» (isotypeswitching). Протеинурия ипочечнаянедостаточность, атакженейросенсорнаяглухота, произошли врезультатепроцессов, инициируемыхотсутствиеколлагена 3 4 5(IV) цепи, аневытекаетнепосредственноиззаотсутствияэтихцепей. α α α
25. 12. 2010 Синдром Альпорта. Клеточнаяимолекулярнаябиология: Как мутацииизменяютфункцииорганеллы : Патология аллельныхвариантов Подавляющее большинствомутаций. COL 4 A 5 этогуаниновыезаменывпервой или второйпозицииглициновыхкодонов. Такиемутации, каксчитается, мешают нормальному сплетениюмутировавшего 5 коллагена(IV)сдругимитипамиα коллагена. Боковойцепинехватаетглицина, иприсутствиегромоздкой аминокислоты вглицинпозициипредположительносоздаетизломилитройная спираль разворачивается. Заменaгицинавколлагене 1(I)цепивызываетα несовершенный остеогенез. Неправильносложеннаятройнаяспиратьколлагена обладает повышеннойвосприимчивостьюкпротеолитическойдеградации. Мутации вгенах. Col. IVAIIICol. IVAIVCol. IVAVIимеюттужеразновидность.
25. 12. 2010 Синдром Альпорта. Спасибозавнимание!