
Лизосомные болезни накопления Максимова Ю.В., НГМУ, г. Новосибирск


Лизосомные болезни накопления Максимова Ю.В., НГМУ, г. Новосибирск

Лизосомы - пузырьки с одномембранной оболочкой диаметром 200-400 мкм
 Основная")
Лизосомы от гр. lysis — «разложение, растворение, распад» и soma — «тело») Основная функция — внутриклеточное переваривание различных химических соединений и клеточных структур. Содержат набор ферментов (кислых гидролаз), которые осуществляют при низких значениях рН гидролитическое расщепление веществ (нуклеиновых кислот, белков, жиров, углеводов).

Лизосомные болезни накопления это группа редких наследственных заболеваний, связанных с недостаточностью определенных метаболических ферментов в лизосомах клеток человека
 Сфинголипидозы Мукополисахаридозы Гликогенозы Муколипидозы Гликопротеинозы")
Лизосомные болезни накопления (ЛБН) Сфинголипидозы Мукополисахаридозы Гликогенозы Муколипидозы Гликопротеинозы Липофусцинозы Другие

Актуальность проблемы ЛБН - Полисистемность поражения органов - Сложность постановки диагноза по клинике - Необходимость дифференциальной диагностики с различными заболеваниями другой этиологии - Возможность точной постановки диагноза методами энзимодиагностики, ДНК-диагностики - Эффективность фермент-заместительной терапии - Возможность профилактики ЛБН методом пренатальной диагностики

Особенности ЛБН Вариабельность сроков манифестации и тяжести Выраженность клинического полиморфизма Прогредиентное течение Единые молекулярные механизмы этиопатогенеза

Сфинголипидозы Болезнь Гоше Болезнь Фабри GM-ганглиозидозы Галактосиалидоз Церамидазы недостаточность Болезнь Нимана-Пика Метахроматическая лейкодистрофия

Болезнь Гоше накопление липидов, обусловленное дефектом гена, ответственного за синтез лизосомального гидролитического фермента β-глюкоцереброзидазы.

Историческая справка 1882 год - французский врач Филипп Чарльз Эрнест Гоше впервые описал болезнь у пациента с увеличенной печенью и селезенкой. 1924 год - немецкий врач выделил жировое вещество из селезенки пациентов с болезнью Гоше. 1934 год - выделен глюкоцереброзид 1964 год - открыт фермент глюкоцереброзидаза

НАСЛЕДСТВЕННОСТЬ

Патогенез болезни Гоше Глюко цереброзид Глюкоза Церамид enzyme Лизосомальная дисфункция Клеточная дисфункция и гибель клеток Поражение органов и систем enzyme Глюко цереброзид

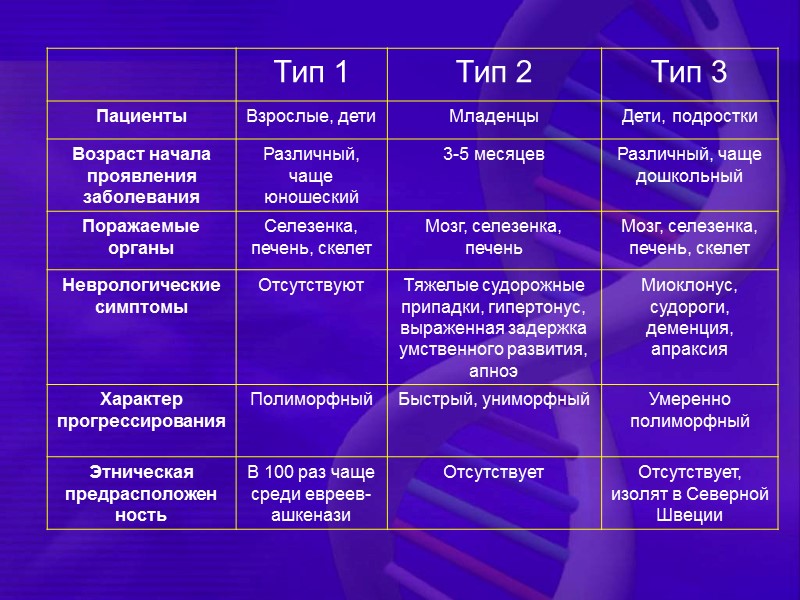
Клинические формы болезни Гоше тип I — хроническая не-нейронопатическая тип II — острая нейронопатическая тип III — подострая нейронопатическая (обе формы встречаются только в детском возрасте)

Клинические проявления болезни Гоше Спленомегалия Гепатомегалия Анемия Тромбоцитопения Кровотечения Кровоподтеки Инфильтрация костного мозга Остопения Остеонекроз Хронические боли в костях Патологические переломы

Клинические проявления Гематологические: Анемия Тромбоцитопения Спонтанный геморрагический синдром: Кровотечения (после инвазивных манипуляций, в т.ч. малых оперативных вмешательств, меноррагии) Кровоподтеки, гематомы

Клинические проявления Висцеральные: Спленомегалия Гепатомегалия
 сильнейшие оссалгии лихорадка локальные")
Клинические проявления Скелетные: Костные кризы подострое начало (12-36 часов) сильнейшие оссалгии лихорадка локальные проявления (резкая болезненность, припухлость, ограничение подвижности сустава) ложный диагноз остеомиелита
 Типичная локализация - дистальный метадиафиз бедренной кости Дефектная")
Колба Эрленмейера (у 25-30% больных) Типичная локализация - дистальный метадиафиз бедренной кости Дефектная реконструкция костной ткани (дисбаланс между оссификацией и резорбцией)

Остеонекроз головки бедренной кости 2 типа: медуллярный субхондральный Ишемия из-за хронического инфаркта кости «кальциевое мыло» Факторы риска спленэктомия мужской пол

Диагностика болезни Гоше 1. Клиническая картина 2. Исследование костного мозга (выявление «пенистых» клеток, диф. диагностика) Диагностика поражений костей (рентгенография, МРТ, денситометрия) 4. Энзимодиагностика - измерение активности β-D-глюкозидазы в лейкоцитах (единственный метод, подтверждающий на 100% болезнь Гоше) Диагностика и доставка анализов крови осуществляется бесплатно (спонсор - компания Genzyme) в Медико-генетическом центре, Москва ОСОБЕННОСТЬ: Молекулярно-генетическая диагностика - отсутствие достоверной связи клинической картины с уровнем активности глюкоцереброзидазы, с генотипом

Диагностика болезни Гоше Лабораторная диагностика тромбоцитопения нормоцитарная анемия лейкопения, панпитопения «пенистые» клетки в пунктате костного мозга, печени Биохимический фенотип Лейкоциты, культура кожных фибробластов: активность лизосомной b-D-глюкозидазы понижена Сыворотка: активность кислой фосфатазы и хитотриозидазы повышена Функциональные исследования УЗИ органов брюшной полости ЭЭГ, КТ/МРТ головного мозга, рентгенография длинных трубчатых костей Клетки Гоше в костном мозге Клетки Гоше в селезенке Клетки Гоше в печени

Дифференциальная диагностика Гемобластозы и лимфопролиферативные заболевания Другие наследственные ферментопатии Нарушение свертываемости крови, гемофилии Анемии Остеомиелит Болезнь Пертеса Костный туберкулез Ревматические заболевания Хронические заболевания печени
 Симптоматическое лечение Генная терапия (в стадии")
Лечение: Фермент-заместительная терапия (Церезим - рекомбинантная глюкоцереброзидаза) Симптоматическое лечение Генная терапия (в стадии разработки)

Способ введения Церезима Режим введения 1 раз в 2 недели Внутривенная инфузия Стандартная доза: 30 ЕД/кг при отсутствии поражения костей 60 ЕД/кг при наличии костных изменений. У 88% больных Гоше костная система вовлечена в процесс, необходимо тщательное диагностическое исследование (рентгенография, МРТ, денситометрия) Пожизненная терапия

Фермент-заместительная терапия Останавливает прогрессирование заболевания Способствует обратному развитию симптомов болезни Гоше Улучшает качество жизни больных Позволяет предотвратить развитие тяжелых осложнений

Мониторинг ферментной терапии болезни Гоше

Спленэктомия «Инвазивный мониторинг»: повторные пункции и биопсии Облучение пораженных костей Энзимодиагностика Заместительная ферментная терапия Неинвазивный мониторинг Лечение осложнений

Болезнь Фабри – Болезнь накопления липидов, обусловленная деффектом гена, ответственного за синтез лизосомного фермента α-галактозидазы A (α-GAL-A).

НАСЛЕДСТВЕННОСТЬ
 и церамид-дигалактозида в стенках сосудов")
ПАТОГЕНЕЗ Дефицит α-GAL-A Накопление церамид-тригексозида (GL-3) и церамид-дигалактозида в стенках сосудов Облитерация (окклюзия) сосудов Нарушению функции клеток и органов
 - манифестирует в детском или юношеском возрасте атипичная (более")
Формы болезни Фабри классическая (тяжелая) - манифестирует в детском или юношеском возрасте атипичная (более легкая) - манифестирует у взрослых или пожилых людей

Фенотип болезни Фабри Увеличены нос, уши, язык, слюнные железы Диспропорциональный рост костей черепа (увеличение скуловых костей, надбровных дуг, затылочных бугров, прогнатия) Увеличение размеров позвонков, расширение грудной клетки, кифоз, лопатообразное увеличение кистей и стоп

Фенотип пациентов с БФ

Проявления и симптомы Ранние ишемические инсульты Гипертрофия левого желудочка Ангиокератомы Акропарастезии Акропарастезии Прогрессирующая почечная недостаточность Гипогидроз
:")
Клинические признаки классической болезни Фабри Ранние проявления (детский и подростковый возраст): Перемежающиеся парестезии и акропарестезии периодические кризы сильных болей в конечностях рецидивирующие подъемы температуры Ангиокератомы Помутнения роговицы и хрусталика Гипогидроз или ангидроз Непереносимость жары и холода Проблемы с желудочно-кишечным трактом Ангиокератомы при болезни Фабри Помутнение роговицы при болезни Фабри
 Сердечно-сосудистые")
Клинические признаки классической болезни Фабри Поздние проявления (молодой и взрослый возраст) Сердечно-сосудистые дисфункции Сосудисто-мозговые осложнения Прогрессирующая почечная недостаточность, ведущая к терминальной стадии поражения почек Легочные осложнения Патология лимфатической системы Конечная стадия болезни Фабри в почках Поражение сердца при болезни Фабри

Клинические признаки атипичной формы болезни Фабри кардиальная или кардио-пульмональная симптоматика: кардиомегалия, гипертрофическая кардиомиопатия, эмболия легочной артерии дистрофия роговицы, реже - ангиокератома акропарестезии

Диагностика болезни Фабри Биохимическая – снижение активности α-ГАЛ в плазме, лейкоцитах, сыворотке, слезах, тканевой биопсии (единственный метод, подтверждающий на 100% болезнь Фабри) Диагностика и доставка анализов крови осуществляется бесплатно (спонсор - компания Genzyme) в Медико-генетическом центре, Москва Морфологическая Молекулярно-генетическая

Болезнь Фабри: Недостаточная осведомленность Если Вы видите 100 больных, нуждающихся в гемодиализе – один из них скорее всего пациент с болезнью Фабри

Лечение болезни Фабри Симптоматическая терапия: для облегчения мучительных акропарестезий: -адренергический блокатор феноксибензамин; сочетание дифенилгидантоина и карбамазепина; аналгетик нейротропин косметическое удаление ангиокератом аргон-лазерной терапией купирование сердечно-сосудистой и цереброваскулярной недостаточности хронический гемодиализ и/или трансплантация почек Фермент-заместительная терапия: препарат "Фабразим" Генотерапия
 – рекомбинантная форма фермента альфа-галактозидазы. Фермент-заместительная терапия болезни Фабри")
Фабразим (агалзидаза бета) – рекомбинантная форма фермента альфа-галактозидазы. Фермент-заместительная терапия болезни Фабри
 Флаконы – 35мг Дозировка 1 мг/кг Вводится в/в капельно Инфузии")
Фабразим (агалзидаза бета) Флаконы – 35мг Дозировка 1 мг/кг Вводится в/в капельно Инфузии 1 раз в 2 недели

Фермент-заместительная терапия Улучшение качества жизни Предотвращение поражения органов-мишеней Улучшение функций пораженных органов Препятствие развитию осложнений

Мукополисахаридозы Наследственные заболевания, связанные с нарушением обмена мукополисахаридов в соединительной ткани в результате неполного распада сульфатированных гликозаминогликанов (ГАГ) в лизосомах клеток.

Классификация мукополисахаридозов

Патогенез МПС I типа Недостаточность фермента -L-идуронидазы Нарушение отщепления терминального остатка идуроновой кислоты Нарушение внутрилизосомной деградации ГАГ Накапление ГАГ в лизосомах

Клинические формы МПС I типа Синдром Гурлер манифестирует на 1 году жизни тяжелая соматическая и неврологическая патология Синдром Гурлер—Шейе манифестирует в 3-8 лет соматическая патология минимальные интеллектуальные расстройства Синдром Шейе Манифестирует в 7-20 лет мягкие проявления фенотипа нормальный интеллект
: Пупочные и")
Клиника МПС I типа Ранние проявления (с рождения до 24 месяцев): Пупочные и паховые грыжи, часто рецидивирующие Костные аномалии черепа, позвоночника (горб), грудной клетки, бедер и конечностей Повторные и затяжные инфекции кишечника и дыхательных путей Задержка развития Помутнение роговицы Гепатоспленомегалия Шум в сердце Утомляемость, летаргия, сонливость Деформация черепа Помутнение роговицы Деформация по типу когтеобразной кисти

Диагностика МПС I типа Биохимический фенотип культура кожных фибробластов, лейкоциты: активность -L-идуронидазы моча: суммарная экскреция гликозаминогликанов , дерматансульфатов , гепарансульфатов Параклипические обследования ЭКГ/ЭхоКГ, КТ/МРТ головного мозга исследование прозрачности глазных сред аудиометрия рентгенологическое исследование скелета

Лечение МПС I типа Симптоматичская терапия: шунтирование для купирования коммуникационной гидроцефалии пересадка роговицы использование слуховых аппаратов терапия отитов и евстахиитов физиотерапия и лечебная гимнастика для борьбы с тугоподвижностью трахеостомия при ночных апноэ Трансплантация костного мозга Фермент-заместительная терапия – АЛЬДУРАЗИМ (ларонидаза)
 Концентрат для приготовления инфузионного раствора , 100 ЕД/мл")
Альдуразим (Aldurazyme) Концентрат для приготовления инфузионного раствора , 100 ЕД/мл
 II тип, дефект кислотной мальтазы (болезнь")
Гликогенозы I тип, дефект глюкозо-6-фосфатазы (болезнь Гирке) II тип, дефект кислотной мальтазы (болезнь Помпе) III тип, нарушение фермента, устраняющего разветвления гликогена (болезнь Кори или болезнь Форбса) IV тип, нарушение процесса ветвления гликогена (болезнь Андерсена) V тип, нарушение мышечной фосфорилазы гликогена (болезнь Мак-Ардля) VI тип, дефект фосфорилазы печени (болезнь Герса) VII тип, дефект фосфофруктокиназы мышц (Болезнь Таруи) IX тип, дефект фосфорилазы киназы XI тип, нарушение транспорта глюкозы (болезнь Фанкони-Бикеля)

Болезнь Помпе Прогрессирующее, мультисистемное, часто фатальное заболевание Впервые описано в 1932 датским патологом J.C. Pompe Также известное как дефицит кислой мальтазы (AMD), болезнь накопления гликогена 2-го типа (GSD-II), или гликогеноз 2- типа Характеризуется прогрессирующей дегенерацией скелетной, дыхательной, и в первую очередь у младенцев, сердечной мускулатуры

Частота симптомов Выраженная гепатомегалия 100 % Нефромегалия 90 % Прогрессирующая печёночная недостаточность 90 % Носовые кровотечения 90 % Задержка физического развития 90 % Анемия 80 % Мышечная гипотония 25 % Кожный ксантоматоз 10 %

Болезнь Помпе, формы течения Детская, начинается в конце периода младенчества или в раннем детстве, быстро прогрессирует. Взрослая, начинается во втором-четвертом десятилетии жизни. Медленное прогрессирование, сердце не поражается.

Генетика и заболеваемость Аутосомно-рецессивный тип наследования Ген GAA расположен 17q25 Больше 200 мутаций в гене GAA www.pompecenter.nl полный список мутаций Имеются слабые корреляции генотип-фенотип 1:40,000

Патогенез болезни Помпе Наследственный дефицит фермента приводит к накоплению гликогена в лизосомах GAA фермент необходим для деградации гликогена в лизосомах

Болезнь Помпе, развитие, клинические проявления Мутации в гене GAA Дефицит фермента GAA Накопление в клетках гликогена Прогрессирование органная недостаточность Патологические изменения мышечных волокон Обездвиженность и лёгочно-сердечная недостаточность Ранняя смертность Клиническая манифестация

Болезнь Помпе, детская форма Kishnani et al. J Pediatr 2006; 148:671-676. Hirschhorn R, Reuser AJJ. New York: McGraw Hill, 2001:3389-3420. With permission from B. Byrne, MD Сердечные изменения Развиваются у 92% детей с 4 месяцев Сердце значительно увеличено Прогрессирует сердечная недостаточность With permission from M. Russell-Taylor, MD Здоровый ребёнок Ребёнок с болезнью Помпе

Сердце Рентген ЭКГ УЗИ Лёгкие Спирометрия Рентген Газовый состав крови Исследование во сне Kishnani et al. Genet Med 2006; 8:267-288. Hirschhorn R, et al. In: The Metabolic and Molecular Bases of Inherited Disease. 2001:3389-3420. Методы, используемые в диагностике

Мышцы Электромиография/проводимость Сила мышц Лабораторные методы Сывороточная креатинкиназа АЛТ, АСТ, ЛДГ Гистология Kishnani et al. Genet Med 2006; 8:267-288. Hirschhorn R, et al. In: The Metabolic and Molecular Bases of Inherited Disease. 2001:3389-3420. An Y, et al. Mol Genet Metab. 2005;85:247-254. Методы, используемые в диагностике

Подтверждение диагноза Определение активности фермента GAA Ранее делалось Биопсия кожи Биопсия мышц Теперь, для определения активности фермента GAA, достаточно одно из Сухое пятно крови Лейкоциты Лимфоциты Анализ мутаций
 Фермент-заместительная терапия Myozyme – первый и единственный специфический препарат для лечения")
Myozyme™*(альфа глюкозидаза) Фермент-заместительная терапия Myozyme – первый и единственный специфический препарат для лечения Pompe disease Производится с использованием технологии рекомбинантной ДНК Заменяет отсутствующий фермент Подобно нативному ферменту GAA, Myozyme расщепляет гликоген * Medicinal product subject to restricted medical prescription

Лечение Рекомендуемая доза 20 mg/kg В/в инфузия 1 раз в 2 недели

Профилактика лизосомных болезней накопления Пренатальная диагностика амниоцентез кордоцентез определение активности ферментов ДНК-диагностика

Благодарю за внимание
29900-b29183d5_maksimova_yu.v._lbn.ppt
- Количество слайдов: 67