
Генетика человека 1. Особенности человека, как объекта исследования


 • Медленная смена")
 • Цитологический (кариотипирование) •")

 – Сдб() 100 - Сдб ()Для оценки наследственности и среды в")

 Сбор сведений о семье(анамнез) (прежде всего сбор сведений")


")




 размеры хромосом,")
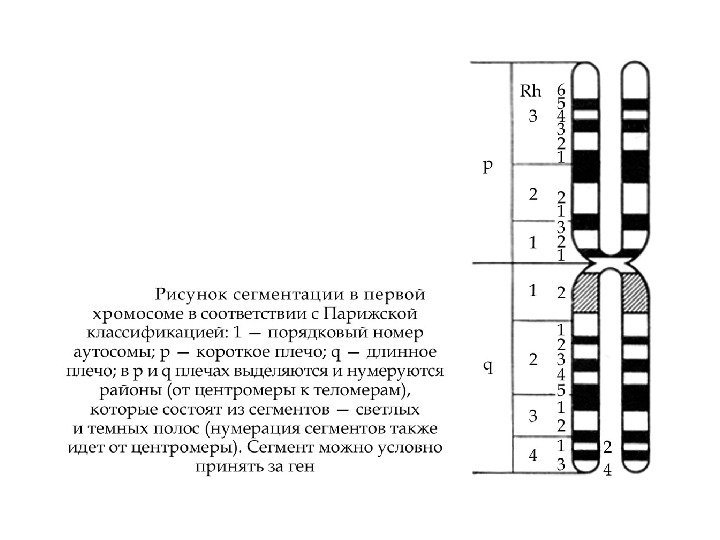
Анализ кариограммы Парижская классификация (1971 год) размеры хромосом положение")
")





 Углеводный обмен (болезнь Тэя-Сакса) Гемоглобинопатии Дефекты биосинтеза гормонов (адреногенитальный")
 Синдром Дауна – 47, ХХ+21, 47,")
 обусловлен делецией длинного плеча 13 -й хромосомы, сегментов 13")
 обусловлен делецией короткого плеча 5 -й хромосомы. Популяционная частота")
; 46, ХY, t(9/22) (хронический миелобластный лейкоз, хронический миелолейкоз, ХМЛ) хромосомная")
 1866 г. , Джон Даун, Англия, педиатр; 1959")
 монголоидный разрез")
 трисомия по хромосоме 18 Джон Эдвардс, Англия, цитогенетик, 1960 г.")
 трисомия по хромосоме 13")
 полисомия по половым хромосомам Гарри Клайнфельтер, американский врач, 1942 г.")
 1925 г. , Николай. Адольфович. Шерешевский, СССР, эндокринолог; 1938")

genetika_cheloveka.ppt.pptx
- Размер: 5.0 Мб
- Автор:
- Количество слайдов: 37
Описание презентации Генетика человека 1. Особенности человека, как объекта исследования по слайдам
Генетика человека 1. Особенности человека, как объекта исследования 2. Методы изучения наследственности человека 3. Наследственные болезни человека
Особенности человека, как объекта исследования • Невозможность искусственного скрещивания (мораль, этика) • Медленная смена поколений, мало потомков • Большое число групп сцепления(23 ♀ , 24 ♂) • Высокая степень полиморфизма (Полиморфизм-различия между гомологичными хромосомами, не оказывающие влияния на фенотип. Обеспечивает уникальность кариотипа каждого человека. ) • Невозможность создания одинаковых условий жизни.
Методы изучения наследственности человека • Клинико-генеалогический (составление и анализ родословных) • Цитологический (кариотипирование) • Близнецовый • Дерматоглифический • Биохимический • Метод гибридизации соматических клеток • Метод рекомбинантной ДНК (анализ и клонирование ДНК, создание геномных библиотек) • Популяционно-статистический • Метод моделирования (создание трансгенных животных моделирующих болезни человека)
Близнецовый метод Изучение закономерностей наследования признаков у моно- и дизиготных близнецов Позволяет: • Оценить степень влияния среды и генотипа на развитие признака в норме и патологии • Выявить наследственный характер признака • Определить пенетрантность аллеля • Оценить эффективность действия на организм некоторых внешних факторов (лекарств, обучения, воспитания) • Процент сходства группы близнецов по изучаемому признаку — КОНКОРДАНТНОСТЬ ; процент различия – ДИСКОРДАНТНОСТЬ • Сравнение конкордантности по данному признаку у монозиготных и дизиготных близнецов помогает более объективно судить о роли генотипа в формировании признака. ))).
Смб (%) – Сдб(%) 100% — Сдб (%)Для оценки наследственности и среды в развитии признака используют формулу Хольцингера: Н- коэффициент наследственности Смб – конкордантность монозиготных близнецов Сдб – конкордантность дизиготных близнецов Н=1 →признак определяется генотипом Н=0 → признак определяется средой 0<Н<1 →влияние и среды и наследственности Близнецовый метод Н=
Клинико-генеалогический метод • Составление и анализ родословных • Выявление типа и характера наследования признака : аутосомно-доминантное, аутосомно-рецессивное, сцепленное с полом, Нетрадиционные типы наследования: цитоплазматическое наследование, однородительские дисомии, болезни экспансии) • Определение пенетрантности и экспрессивности гена • Вычисление генетического риска
Клинико-генеалогический метод. СОСТАВЛЕНИЕ РОДОСЛОВНОЙ: 1) Сбор сведений о семье(анамнез) (прежде всего сбор сведений о пробанде — индивиде, который является предметом интереса исследователя (врача, педагога). Чаще всего это больной или носитель изучаемого признака. )) Дети одной родительской пары (братья и сестры) называются сибсами. Если сибсы имеют только одного родителя, они называются полусибсами. Различают: единоутробных сибсов (общая мать); единокровных сибсов (общий отец). Обычно родословная собирается в связи с изучением одного или нескольких заболеваний (признаков). Чем больше поколений вовлекается в родословную, тем больше информации она может содержать. Для составления родословной используют определенные символы: 2) Построение родословной 3) Анализ родословной и выводы
Родословная с концентрическим расположением поколений
Родословная семьи с брахидактилией (недоразвитость дистальных фаланг пальцев, приводящая к короткопалости)
Цитологический метод • Кариотипирование – анализ числа и морфологии хромосом клетки на стадии метафазы митоза (определение пола, диагностика хромосомных болезней, изучение мутагенеза), составление кариограммы и идеограммы. • Изучение полового хроматина (экспресс-диагностика половой принадлежности, быстрое определение хромосомных болезней, связанных с изменением числа хромосом по количеству телец Барра) • Метод флуоресцентной in situ гибридизации (FISH) : локализация генов в хромосомах, выявление хромосомных аномалий при пренатальной диагностике, ген. Тестирование эмбрионов при ЭКО)
Тельце Барра
Кариограмма человека. А- женщины, Б-мужчины
Идеограмма нормального кариотипа человека
Анализ кариограммы Денверская классификация хромосом (1960 -1966 г. г. ) размеры хромосом, положение центромеры
Парижская классификация хромосом (1971 г)Анализ кариограммы Парижская классификация (1971 год) размеры хромосом положение центромеры дифференциальное окрашивание
Метод флуоресцентной in situ гибридизации (FISH)
Метафазная пластинка и интерфазные ядра из лимфоцитов пациента с кариотипом 47, ХХУ после 5 -цветной флуоресцентной гибридизации in situ. Участки хромосом маркированы цветами: 13 хр. – красным, 21 хр. — зеленым, 18 хр. — голубым, Х хр. -синим, Yхр. — желтым. Метод флуоресцентной in situ гибридизации (FISH)
Дерматоглифический метод • Изучение рисунка на ладонях, подошвах и пальцах. (дактилоскопия — пальцы, пальмоскопия — ладони, плантоскопия – ступни ног ) • Используется в медико-генетических консультациях для уточнения клинического диагноза, • дифференциации различных форм заболеваний, • для выяснения носителей мутантных аллелей. Синдром Дауна (Трисомия 21) — наличие «Обезьяньей линии» на обеих руках, учащение узоров на подушечке под большим пальцем. Синдром Клайнфельтера (47, ХХУ) — учащенный узор на подушечке под большим пальцем изменение дерматоглифики обнаружены при множественных, врожденных пороках развития ЦНС, сердечно – сосудистой системы, желудочно-кишечного тракта.
Биохимический метод • Обнаружение биологически активных соединений и их метаболитов в жидкостях организма • Позволяет выявить нарушения в обмене веществ, вызванные изменением генов и, как следствие, изменением активности различных ферментов. (фенилкетонурия, серповидно-клеточная анемия) • С помощью биохимических методов открыто около 500 молекулярных болезней, являющихся следствием проявления мутантных генов. К наследственным болезням обмена веществ относят болезни углеводного обмена (сахарный диабет), обмена аминокислот, липидов, минералов и др. . • Биохимический скрининг по определению содержания альфафетопртеина (АФП) и хорионического гонадотропина (ХГ) в крови беременных на сроке 15 -18 недель с целью выявления женщин, имеющих повышенный риск рождения ребенка с хромосомными болезнями (синдром Дауна и Эдвардса), а так же с ДЗНТ (дефекты заращения нервной трубки). При синдроме Дауна у плода уровень АФП в крови беременной ниже в 2 и более раз. При наличии синдрома Эдвардса у плода резко снижен уровень ХГ. При анэнцефалии, открытой spina bifida, дефектах передней брюшной стенки (омфалоцеле, гастрошизисе, гидронефрозе и т. д. ) уровень АФП в 3 и более раз повышен по сравнению с нормой. • Трудоемкость • Требуется специальное оборудование
Метод гибридизации соматических клеток • основан на размножении соматических клеток в искусственных условиях • используются культуры соматических клеток , полученные из материала биопсий (кровь, кожа, опухолевая ткань, ткань эмбриона) для генетических исследований человека. • дает возможность анализировать генетические процессы в отдельных клетках и использовать их для изучения генетических закономерностей целостного организма. • позволяет изучать механизмы первичного действия и взаимодействия генов, регуляцию генной активности.
Популяционно-статистический метод Позволяет изучить : • распространение отдельных генов и признаков(в том числе и заболеваний) в человеческих популяциях, • соотношение между частотой гомозигот и гетерозигот • роль наследственности и среды в возникновении болезней, особенно с наследственной предрасположенностью. Производится: выборочное исследование части популяции: изучение архивов больниц, родильных домов, проводится опрос путем анкетирования Используются: методы статистического анализа (закон Г. Харди и В. Вайнберга и ряд других специальных математических методов) для определения частоты генов в различных группах населения, частоты гетерозиготных носителей ряда наследственных аномалий и болезней. Например, гомозиготы по гену Нb. S в Беларуссии практически не встречаются, а в странах Западной Африки частота их варьирует от 25% в Камеруне до 40% в Танзании. Изучение распространения генов среди населения различных географических зон (геногеография) дает возможность установить центры происхождения различных этнических групп и их миграции, определить степень риска появления наследственных болезней у отдельных индивидуумов.
Аминокислотный обмен (альбинизм, фенилкетонурия) Углеводный обмен (болезнь Тэя-Сакса) Гемоглобинопатии Дефекты биосинтеза гормонов (адреногенитальный синдром, синдром тестикулярной феминизации) Мышечные дистрофии, муковисцидоз Наследственные болезни человека ХРОМОСОМНЫЕ МУЛЬТИФАКТОРИАЛЬНЫЕ БОЛЕЗНИ С НЕТРАДИЦИОННЫМ ТИПОМ НАСЛЕДОВАНИЯ • Разнообразие (различные мутации в одном гене) • Различные клинические проявления • Минимальное влияние внешней среды ГЕННЫЕ Патологические реакции на факторы внешней среды: Лекарства, пищевые добавки, тепло, холод, солнечный свет… Болезни предрасположенности (псориаз, шизофрения, сердечно-сосудистые з-я…) Митохондриальные болезни Болезни экспансии (хорея Гентингтона, с-м ломкой хромосомы=с-м Мартина-Белл) Генный импринтинг (синдром Прадера-Вилли, 15 Хр. )Геномные гетероплоидии: Полиплоидии (3 n=69, 4 n=92) Анеуплоидии – моносомии (2 n-1=45) нуллисомии (2 n-2=44), трисомии (2 n+1=47), полисомии(2 n+2=48, 2 n+3=49 для Х, Y) Структурные изменения: Внутрихромосомные- делеции, инверсии, дупликации… Межхромосомные транслокации- Сбалансированные(реципрокные) Несбалансированные (нереципрокные) Робертсоновские транслокации
Хромосомные болезни человека Изменение числа аутосом (аутосомные мутации) Синдром Дауна – 47, ХХ+21, 47, ХY+21 Синдром Эдвардса – 47, ХХ+18; 47, ХY+18 Синдром Патау — 47, ХХ+13; 47, ХY+13 Изменение числа половых хромосом (генеративные мутации) Синдром Шерешевского-Тернера – 45, ХO Синдром Клайнфельтера- 47, ХХY ( 48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY; 49 XXXYY) Полисомия Х хромосомы- Трисомия по Х -47, ХХХ (48, ХХХХ; 49, ХХХХХ…) Нормальный кариотип — 46, ХХ женщина; 46, ХY мужчина Структурные перестройки Хронический миелолейкоз-46, ХХ, t(9/22); 46, ХY, t(9/22)- реципрокная перестройка Транслокационный С. Дауна-46, ХХ, t(15/21); 46, ХY, t(15/21) – Робертсоновская транслокация Синдром Орбели — 46, ХХ, del(13 q-); 46, ХY, del(13 q-) Синдром «Кошачьего крика» синдром Лежёна -46, ХХ, del(5 p-); 46, ХY, del(5 p-) делеции
Синдром Орбели (13 q-) обусловлен делецией длинного плеча 13 -й хромосомы, сегментов 13 q 22 -q 31. Популяционная частота синдрома не установлена. Дети с синдромом Орбели рождаются с низким весом (2200 г). Клинически синдром проявляется аномалиями развития всех систем организма. Характерны микроцефалия, отсутствие носовой вырезки (лоб непосредственно переходит в нос), эпикант, антимонголоидный разрез глаз, широкая спинка носа, высокое нёбо, низко расположенные деформированные ушные раковины. Отмечаются поражения: • глаз (микрофтальмия, иногда анофтальмия, косоглазие, катаракта, ретинобластома), • опорно-двигательного аппарата (короткая шея, гипо- или аплазия первого пальца кисти и пяточной кости, синдактилии кистей и стоп), • атрезии прямой кишки и заднепроходного отверстия. • Часты пороки развития сердца, почек, головного мозга. Для всех детей с синдромом Орбели характерна глубокая олигофрения, возможны потеря сознания, судороги. Большинство больных с синдромом 13 q- погибают на 1 -м году жизни.
Синдром кошачьего крика (5 р-) обусловлен делецией короткого плеча 5 -й хромосомы. Популяционная частота синдрома -примерно 1: 45 000. Для данного синдрома наиболее характерны : • специфический плач, напоминающий кошачье мяуканье , • лунообразное лицо, • мышечная гипотония, • умственное и физическое недоразвитие, микроцефалия, • низко расположенные, иногда деформированные ушные раковины, эпикант, антимонголоидный разрез глазных щелей, косоглазие. Иногда наблюдаются атрофия зрительного нерва и очаги депигментации сетчатки. Как правило, выявляются пороки сердца. Наиболее постоянный признак синдрома — «кошачий крик» — обусловлен изменениями гортани: сужением, мягкостью хрящей, отечностью или необычной складчатостью слизистой оболочки, уменьшением надгортанника. Изменения других органов и систем неспецифичны. Продолжительность жизни у больных с этим синдромом значительно снижена, только около 14% из них переживают возраст 10 лет.
Хронический миелолейкоз-46, ХХ, t(9/22); 46, ХY, t(9/22) (хронический миелобластный лейкоз, хронический миелолейкоз, ХМЛ) хромосомная транслокация, приводящая к образованию филадельфийской хромосомы , получившей своё название по месту работы её первооткрывателей, Питера Ноуелла и Дэвида Хангерфорда, 1960 г. Филадельфиия) • болезнь, при которой наблюдается избыточное образование гранулоцитов в костном мозге и повышенное накопление в крови как самих этих клеток, так и их предшественников. Слово «хронический» в названии болезни означает, что процесс развивается сравнительно медленно, в отличие от острого лейкоза, а «миелоидный» означает, что в процесс вовлечены клетки миелоидной (а не лимфоидной) линии кроветворения.
Синдром Дауна (47, ХХ/XY +21) 1866 г. , Джон Даун, Англия, педиатр; 1959 г. Жером Лежен, Франция, генетик –хромосомная природа 1: 700 -800 новорожденных Причины: • Трисомия по 21 хромосоме (нерасхождение в І делении Мейоза, риск повышается с возрастом родителей; ~ 94%) • Робертсоновские транслокации 14/21; 15/21 хромосом (семейная форма, транслокация возникает у родителей, не отражаясь на фенотипе, возраст родителей не имеет значения; ~4%) • Мозаицизм ( нерасхождение возникает в клетке зародыша на ранних стадиях его развития, нарушение кариотипа затрагивает только некоторые ткани и органы (46, XX/47, XX, +21); ~2%)
Симптомы: плоское лицо , плоский затылок брахицефалия (аномальное укорочение черепа) монголоидный разрез глаз , эпикантус ( вертикальная кожная складка, прикрывающая медиальный угол глазной щели) гиперподвижность суставов поперечная ладонная складка «обезьянья» брахимезофалангия (укорочение всех пальцев за счёт недоразвития средних фаланг) Сопутствующие заболевания: • врожденные пороки сердца, • болезнь Альцгеймера, • острый миелоидный лейкоз • ослабленный иммунитет • нарушения пищеварения Широкая посадка между большим и следующим пальцем ноги слабый тонус мышц повышенное содержание пуринов когнитивные нарушения раннее старение
Синдром Эдвардса(47, ХХ/ХY+18) трисомия по хромосоме 18 Джон Эдвардс, Англия, цитогенетик, 1960 г. 1: 7000 новорожденных, ♀ в 3 раза больше ♂ Причины: • Трисомия по 18 хромосоме (нерасхождение хромосом в Мейозе, риск повышается с возрастом матери; ~ 90%) • Мозаицизм ( нерасхождение возникает в клетке зародыша на ранних стадиях его развития, нарушение кариотипа затрагивает только некоторые ткани и органы (46, XX/47, XX, +18); ~10%) Симптомы: Низкая масса при рождении Сниженная двигательная активность аномалии мозгового и лицевого черепа глазные щели узкие и короткие ушные раковины деформированы аномальное развитие стопы пороки сердца и крупных сосудов Продолжительность жизни : 60 % детей доживают до 3 месяцев, до года 5 -10 %. Основная причина смерти — остановка дыхания и нарушения работы сердца и почек. Оставшиеся в живых — глубокие олигофрены
Синдром Патау(47, ХХ/ХY +13) трисомия по хромосоме 13 Клаус Патау, американский генетик, 1960 1: 6000 -9000 новорожденных, ♀=♂ Причины: • Трисомия по 13 хромосоме (нерасхождение хромосом в Мейозе, риск повышается с возрастом матери; ~ 85%) • Робертсоновские транслокации 13/14 хромосом • Мозаицизм ( нерасхождение возникает в клетке зародыша на ранних стадиях его развития, нарушение кариотипа затрагивает только некоторые ткани и органы (46, XX/47, XX, +13); редко) Симптомы: Низкая масса при рождении Сниженная двигательная активность аномалии мозгового и лицевого черепа Скошенный лоб Запавшая переносица Расщелины губы и неба полидактилия пороки сердца и крупных сосудов изменения поджелудочной железы, добавочные селезёнки, пороки развития половых органов Продолжительность жизни : 95% детей доживают до года, ( до 10 лет 2 — 3 % детей). Оставшиеся в живых — глубокие олигофрены
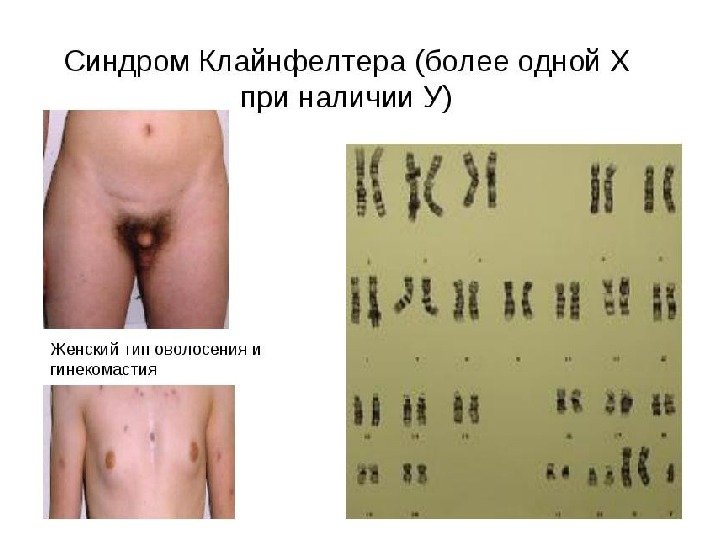
Синдром Клайнфельтера (47, ХХY) полисомия по половым хромосомам Гарри Клайнфельтер, американский врач, 1942 г. 1: 500 -600 новорожденных мальчиков Варианты : 48 ХХХY, 49 ХХХYY…. Причины: патология мейоза — нерасхождение хромосом при овогенезе ~70%, при сперматогенезе ~30% Факторы риска — возраст матери Клинические проявления диспропорциональное строение тела – высокая талия, длинные конечности Гинекомастия (увеличение грудных желез) ожирение и оволосение по женскому типу недоразвитие сперматозоидов, бесплодие Психологические особенности- манерность, болтливость Умственное развитие-от нормы до легкой степени УО (? )
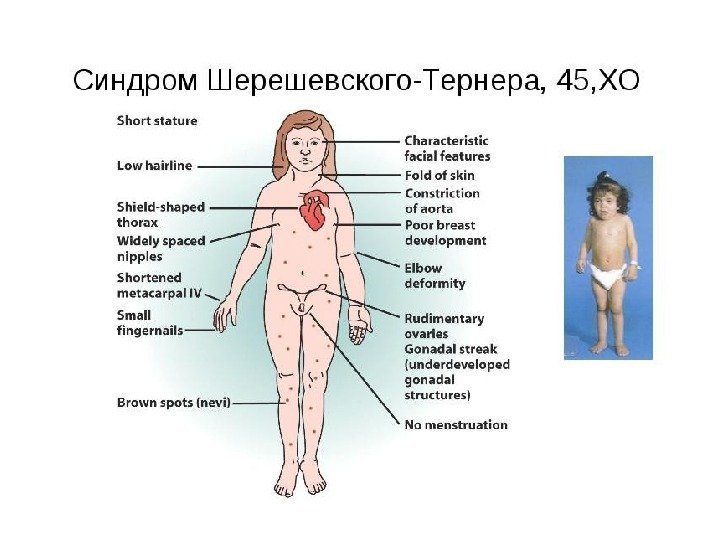
Синдром Шерешевского-Тернера(45, ХО) 1925 г. , Николай. Адольфович. Шерешевский, СССР, эндокринолог; 1938 г. , Генри Тёрнер, США — врач – триада симптомов 1959 г. , Ч. Форд – раскрыта этиология заболевания (моносомия по X-хромосоме) 1: 3000 девочек Причины: нарушения расхождения хромосом при гаметогенезе Симптомы: • половой инфантилизм • кожные крыловидные складки на боковых поверхностях шеи • деформация локтевых суставов • Низкий рост (135 -145 см) • Интеллект-норма (характерен психический инфантилизм с эйфорией)
n+1 n-