
Drug development process I wish














 – declared that the only difference")



 that could puncture the uterus")




 new insights into a disease")


 studies using component of organism Examples: • Cells derived from")

 organism: Metabolic profile")




 Who qualifies to participate; b) How many people will be part of the")
 Project Manager; b) Medical Officer; c) Statistician;")

 people with disease is recruited.")









 involves a complex interaction")

drug_discovery_and_approval_process1.pptx
- Размер: 10.8 Мб
- Автор:
- Количество слайдов: 50
Описание презентации Drug development process I wish по слайдам
Drug development process
I wish my job was sleeping
Mom, I’m coming back from work. What should I get from the store? Cereal? Peanut butter? Some milk? Buy your own apartment, my dear….
What is a main responsibility of pharmaceutical industry?
Human civilization achievements
1. Choosehealthy foods, 2. Fit more exercise and physical activity into your daily routine, 3. Practice good hygiene, 4. Avoid unhealthy habits.
1. There a lot of diseases which can be cured only by using medications, 2. Many of well established drugs in fact have different side effects; 3. There are some incurable or hardly curable diseases; 4. A mutation of pathogenic organisms – lead to “new” diseases (cold and influenza viruses are examples of rapidlymutating pathogens)
What is a main responsibility of pharmaceutical industry?
The pharmaceutical industry is responsible for the development, production, marketing of medications.
Some statistics In 2010 the costs of development a new drug were between $4 billion to $11 billion per drug.
TWO CRUCIAL TERMS IN DRUG DEVELOPMENT: — EFFECTIVENESS AND —
The basis of the world’s pharmacology before 20 th century : — inherited knowledge, — trial and error, — mystical theories.
Swiss physician, and chemist Paracelsus (1493 -1541) – declared that the only difference between a medicine and a poison was in the dose.
150 years ago the average life expectancy in the World was 30 -35 years The vast majority of deaths before the mid-20 th century were caused by microbes; Use of unchlorinated, unfiltered water was attributed to 1/2 of the overall mortality, 2/3 of child mortality, 3/4 of infant mortality SOME STATISTICS
«William Radam’s Microbe Killer” and “Benjamin Bye’s Soothing Balmy Oils to cure cancer, “ Unpasteurized milk, Cows weren’t tested for TB, Opium, morphine, heroin, and cocaine – no restrictions or labeling. At the beginning of 20 th century there were no regulation in drug production and marketing.
The first significant consumer protection law in US: Prohibited foreign and interstate traffic in adulterated or mislabeled food and drug product; Required that active ingredients be placed on the label of a drug’s packaging; Required that drugs do not fall below purity levels established by existent standards; Allowed for seizure and criminal penalties. • Did not address : – Food or drug standards; – False advertising; – Inspection of food and drug facilities; – regulation of new drug safety The Pure Food and Drug Act of
◦ A womb supporter (also used as a contraceptive) that could puncture the uterus if inserted incorrectly; ◦ A weight-loss drug that caused death; ◦ A hair remover that caused baldness, even if not used on the head; ◦ Lotions and creams that could cause mercury poisoning; ◦ An eyelash dye that blinded women; ◦ Hair dyes that could cause lead poisoning. CAUSE no regulations to ensure the safety of products. “ America’s Chamber of Horrors” (1933)
1937 ELIXIR SULFANILAMIDE TRAGEDY PRESCRIBED as to treat streptococcal infections CONTAINED antifreeze Diethylene glycol as solvent RESULTS 107 people, mostly children, die CAUSE – no legal requirement to list inactive ingredients on label RESOLUTION Federal Food, Drug & Cosmetics Act of 1938 – company were required to prove their products are safe before marketing them
PRESCRIBED as sleeping pills and to ease morning sickness in pregnancy. Declared as “completely safe” for everyone, including mother and child. RESULTS Peripheral neuritis –nerve disorder – Birth defects –deafness, blindness, disfigurement, cleft palette, internal defects, phocomelia (the shortening or absence of limbs – about 10, 000 cases) CAUSE — faulty of preclinical and clinical procedures; — no centralized regulatory organization. RESOLUTION catalyzes the beginnings of the rigorous drug approval and monitoring systems. THE THALIDOMIDE TRAGEDY in 1960’s
Evolution of regulatory mechanism in drug production and marketing in US.
Present Timeline from Molecule to Market
Classical pharmacology; Reversed pharmacology : a) new insights into a disease mechanism; b) identification of molecular target; c) ligand- or structure-based drug design (computer-aided drug design, etc); Existing treatments that have unanticipated effects (Viagra); New technologies , which provide new ways to target medical products to specific sites within the body or to manipulate genetic material (nanotechnologies, etc). Thousands of compounds may be R & D STEP 1 a: DRUG DISCOVERY Eureka! !!
MAJOR CLASSES OF DRUG TARGETS
STEP 1 b: PRECLINICAL DEVELOPMENT OF NEW LEADS Give an information on: How it is A bsorbed, D istributed , M etabolized, and E xcreted; Its mechanisms of action and potential benefits; The best way to give the drug; The best dosage; Side effects; How it affects different groups of people; How it interacts with other drugs and treatments; Its effectiveness as compared with similar drugs. In vivo animal and in vitro animal or human cell studies, HTS
(in glass) studies using component of organism Examples: • Cells derived from multicellular organisms; • Subcellular components (ribosomes, mitochondria); • Cellular/ subcellular extracts (wheat germ); • Purified molecules (DNA, RNA). IN VITRO STUDIES
Advantages : Studies can be completed in short period of time; Permits an enormous level of simplification of the system (eg. , allow to control extracellular environment); Use of animals reduced. Disadvantages: Removal of components of organism (eg. cells) from their in vivo environment can influence their function (hormones, factors, other cells); “ Artificial” microenvironment could influence the cellular function.
The experimentation using a whole, living organism. In control (healthy) organism: Metabolic profile Toxicology Drug interaction Animal models of disease or injury (eg. , TBI): Test conditions involving induced or genetic disease or injury similar to human conditions; Can predict human toxicity in 71 % of the cases. IN VIVO STUDIES
Other Advantages Reduced harm to humans; Behavioral studies (investigate behavioral outcomes of drugs; observe mental disorders); Functional Imaging (detecting or measuring changes in metabolism, blood flow, and absorption — CT, MRI). Disadvantages Increased harm to animals involved; Expensive; Legal paperwork; Limited by the fact that animals are not humans.
A method which use robotics, data processing and control software, liquid handling devices, and sensitive detectors to quickly conduct millions of chemical, genetic, or pharmacological tests. The key labware — the microplate. ADVANTAGES: a) screening a biological target against thousands of chemical leads; b) automating and miniaturizing the assay process; c) decreasing the time (1000 times faster) of screening and its cost (at 1 -millionth cost) comparatively to the conventional techniques. DISADVANTAGES: b) Expensive equipment; c) Sophisticated process (require the expensive support) HIGH-THROUGHPUT SCREENING
Present Timeline from Molecule to Market
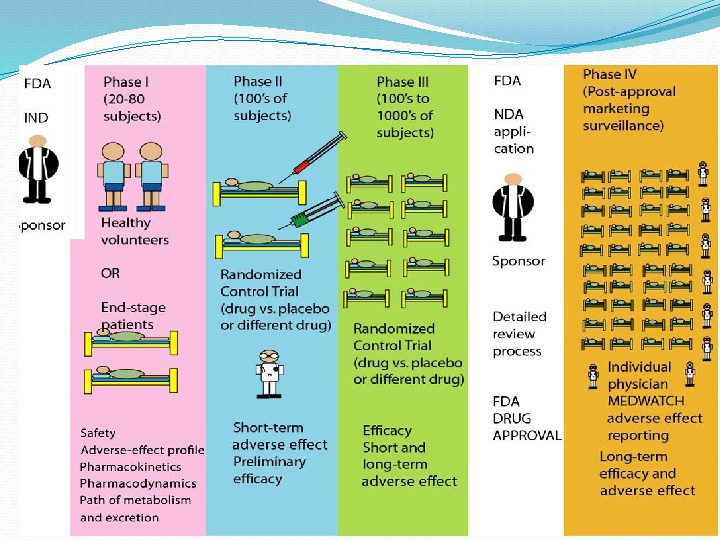
GOAL – to determine the safety and effectiveness of medications, intended for human use. Very first step — the Investigational New Drug Application is a submission to FDA requesting permission to initiate a clinical study of a new drug product. Information included: a) Animal study data and toxicity data; b) Manufacturing information; c) Protocols for clinical studies; d) Data from any prior human research; e) Information about the investigator. STEP 3: CLINICAL RESEARCH in DRUG DISCOVERY
a) Who qualifies to participate; b) How many people will be part of the study; c) How long the study will last; d) Whethere will be a control group and other ways to limit research bias; e) How the drug will be given to patients and at what dosage; f) What assessments will be conducted, when, and what data will be collected; g) How the data will be reviewed analyzed (sample size, statistical analysis, etc) CLINICAL TRIAL DESIGN
The FDA review team includes: a) Project Manager; b) Medical Officer; c) Statistician; d) Pharmacologist; e) Chemist; f) Microbiologist, in case of antimicrobial product. ) 30 days to review after Investigational New Drug Application submission; ) approve, reject or delay the process. The process protects volunteers who participate in clinical trials from unreasonable and significant risk in clinical trials.
Aims to evaluate Safety; Tolerability; Pharmacokinetics ( ADME studies – A bsorption, D istribution, M etabolism, E xcretion); Pharmacodynamics; Dose Ranging (Dose Escalation): 1) Phase 1 a Single Ascending Dose (SAD) or Maximum Tolerated Dose, 2) Phase 1 b Multiple Ascending Dose (MAD); 3) Food Effect. PHASE I TRIAL
A larger group (e. g. 100– 300/group) people with disease is recruited. Aims to evaluate short-term: Safety; Efficacy; Drug toxicity; Drug interaction; Compare to other Drugs PHASE II TRIAL
PHASE III TRIAL The final stage of testing in human subjects prior to licensing. Most expensive; Time consuming; A large group people with disease recruited; Often randomised, multi-centre studies. Aims to evaluate short- and long-term • Safety/Tolerability; • Efficacy
REGULATORY AUTHORITIES suggest new pharmaceutical for sale and marketing. US Food and drugs Agency (FDA) European Medicines Agency (EMA) State Administration of Ukraine on Medicinal Products (SAUMP)
PHASE IV TRIAL Post-Marketing Surveillance after the drug is approved to be sold and is used in medical practice. Aims to detect Rare or long-term adverse effects; Adverse Drug Reaction Monitoring; Drug Interactions.
DEVELOPMENT OF ORPHAN DRUGS
‘ Orphan drugs’ are drugs intended to treat rare diseases A rare disease is any disease that affects a small percentage of the population. Authorities in different countries or regions use different cut-off values to define the disease as ‘Rare’ : WHO — an incidence of 0. 65 -1/1000 US — < 1/1500 patients EU — < 1/2, 000 Japan — < 50, 000 Australia — <
Cystic fibrosis , affects the respiratory and digestive systems; Huntington’s disease , affects the brain and nervous system; Muscular dystrophies , which affect the muscles; All forms of cancer in children are generally considered rare. EXAMPLES OF RARE DISEASES
About 80 % of rare diseases have a genetic component; The European Organization for Rare Diseases estimates that as many as 5, 000 to 7, 000 distinct rare diseases exist ; More the 30 mln of the population of the EU and an 25 — 30 mln in US are affected. Only about 400 rare diseases have therapies STATISTICS
CHALLENGES IN THE ORPHAN DRUGS DEVELOPMENT Difficulties to attract public attention and receive funding for research and development; Insufficient numbers of research participants for clinical studies (sample size is too small); Lack of knowledge for many rare diseases; Lack of adequate expertise and review by authorities; High price of «orphan drugs»
• Funding towards investigation; • Tax credit for clinical research; • Waiver of fees for new drug application; • Accelerated approval or fast track or priority review; • Enhanced patent protection. The Orphan Drug Act (ODA) (1983) and Rare Diseases Act in (2003) US, is meant to encourage pharmaceutical companies to develop drugs for diseases that have a small market INITIATIVES TO STIMULATE ORPHAN DRUG DEVELOPMENT
A success in drug discovery (commercial, or a public health) involves a complex interaction between a) investors, b) industry, c) academia, d) patent laws, e) regulatory exclusivity, f) marketing. For rare disorders which have no large commercial success or public health effect, the Orphan Drug. Funding programs gives a hope for people with these disorders to use some pharmacotherapeuticadvances. SUMMARY
Thank you!