
Диффузные болезни соединительной ткани Общее понятие о ДБСТ


Диффузные болезни соединительной ткани

Общее понятие о ДБСТ прежнее название этой группы заболеваний – «коллагенозы» групповое классификационное понятие, объединяющее ряд системных воспалительных ревматических заболеваний, каждое из которых имеет характерные клинические и нередко иммунологические диагностические признаки.

Диффузные болезни соединительной ткани Согласно современным классификациям ревматических заболеваний, в этот подкласс включены: 1. системная красная волчанка; 2. склеродермия; 3. дерматомиозит; 4. диффузный эозинофильный фасциит; 5. ревматическая полимиалгия; 6. рецидивирующий полихондрит и панникулит; 7. синдром Шегрена; 8. смешанное заболевание соединительной ткани (синдром Шарпа) и перекрестные формы системных заболеваний соединительной ткани

Диффузные болезни соединительной ткани Классификация по МКБ КОД ПО МКБ10: M32 Системная красная волчанка М34 Системный склероз М33 Дерматомиозит М35 Другие системные поражения соединительной ткани М35.3 Ревматическая полимиалгия М 94.1 Рецидивирующий полихондрит М35.0 Сухой синдром М35.1 Другие перекрестные синдромы

Особенности ДБСТ сходство клинических проявлений, особенно в ранней стадии болезни, что создает трудности для их распознавания; прогрессирующее и рецидивирующее течение заболеваний; общие клинические симптомы: артриты (полиартриты), миозиты (миалгии), реже рецидивирующие серозиты, разнообразные висцериты, включая поражение почек и генерализованный васкулит, лимфаденопатию, поражение цнс; сходно изменяются лабораторные показатели воспалительной активности (соэ, уровень альфа2-глобулинов, серомукоида и др.); особенностью патогенеза всех ДБСТ является гиперпродукция аутоантител к компонентам ядра и цитоплазмы клеток (антитела к днк, рнк, рнп, антинуклеарный фактор), fc-рецепторам иммуноглобулинов (ревматоидные факторы), компонентам свертывающей системы (волчаночный антикоагулянт), фосфолипидам (кардиолипин) в сочетании с органоспецифическими аутоиммунными синдромами (например, тиреоидит хашимото, гемолитическая анемия, тромбоцитопеническая пурпура). Аутоиммунные нарушения, как правило, сочетаются с избыточным образованием циркулирующих иммунных комплексов, отложением их в органах-мишенях (кожа, почки, легкие), сосудах и развитием системного иммуноопосредованного воспаления.

Особенности течения ДБСТ у детей У детей вследствие анатомо-физиологических особенностей все диффузные болезни соединительной ткани протекают тяжелее, чем у взрослых. Этим болезням свойственно острое, бурное развитие с быстрым формированием полисистемного патологического процесса с выраженными и распостраненными сосудистыми реакциями, экссудативным компонентом воспаления при большой вероятности рецидивов и дальнейшего прогрессирования. Заболевают чаще девочки, преимущественно младшего школьного и препубертатного возраста.

Этиология и патогенез ДБСТ Этиология ДБСТ неизвестна, хотя предполагается роль некоторых вирусов. Особенностью ДБСТ является мультифакторный тип предрасположения с определенной ролью иммуногенетических факторов (генов локуса d/dr), определяющих тенденцию к возникновению аутоиммунных и иммунокомплексных процессов. По-видимому, в качестве этиологических можно рассматривать половые факторы (гиперэстрогенемия) и воздействия окружающей среды, такие, как инфекции, инсоляция, охлаждение, стрессы психические и физические, несбалансированное питание, реализация которых осуществляется в связи с семейно-генетическим предрасположением к аутоиммунным реакциям и гиперэстрогенемии, чем обусловлено развитие всех этих заболеваний преимущественно у женщин. В основе пато- и морфо- генеза лежат гуморальные и клеточные иммунопато-логические процессы.

СИСТЕМНАЯ КРАСНАЯ ВОЛЧАНКА системное аутоиммунное заболевание неизвестной этиологии, характеризующееся гиперпродукцией органонеспецифических аутоантител к различным компонентам клеточного ядра с развитием иммуновоспалительного повреждения тканей и внутренних органов.

Эпидемиология СКВ Заболеваемость СКВ колеблется от 4 до 250 случаев на 100 000 населения. Пик заболеваемости приходится на 15–25 лет. Женщины страдают в 8-10 раз чаще мужчин. У детей соотношение девочек к мальчикам меньше - 3:1. В препубертатном возрасте частота заболевших мальчиков и девочек примерно одинакова. СКВ наиболее часто развивается в репродуктивном возрасте, во время беременности и в послеродовом периоде. Отмечено увеличение частоты и тяжести течения болезни среди лиц чёрной расы, пуэрториканцев, китайцев. Смертность при СКВ в 3 раза выше, чем в популяции.
; Наследственная предрасположенность; Нарушения гормональной регуляции; Полиэтиологическая")
Этиология СКВ Вирусная и/или бактериальная инфекция (вирус Эпштейна-Барр); Наследственная предрасположенность; Нарушения гормональной регуляции; Полиэтиологическая концепция

Патогенез СКВ Патогенез СКВ определяют два тесно взаимосвязанных процесса: 1. на ранней стадии заболевания преобладает поликлональная (B-клеточная) активация иммунитета, в дальнейшем — антигенспецифические (T-клеточные) иммунные реакции; 2. фундаментальное иммунное нарушение, лежащее в основе СКВ — врождённые или индуцированные дефекты программированной гибели клеток (апоптоза).

Классификация СКВ Выделяют: 1.Острое течение (характерно быстрое развитие мультиорганных проявлений, включая поражение почек и ЦНС, и высокая иммунологическая активность) 2.Подострое течение (в дебюте наблюдаются конституциональные симптомы, неспецифическое поражение кожи и суставов. Заболевание протекает волнообразно, с периодическим возникновением обострений и развитием полиорганной симптоматики в течение 2–3 лет с момента появления первых симптомов) 3.Первичнохронический вариант течения (свойственно длительное превалирование одного или нескольких симптомов: дискоидных высыпаний, синдрома Рейно, артрита, судорожного синдрома, гематологических нарушений, синдрома Шегрена)

Классификация СКВ Клинико-иммунологические варианты СКВ: СКВ с дебютом в детском и подростковом возрасте; «СКВ у пожилых»; СКВ у мужчин; Подострая кожная красная волчанка; Вторичный АФС; Синдром неонатальной волчанки

Клиническая картина СКВ 1.КОНСТИТУЦИОНАЛЬНЫЕ НАРУШЕНИЯ: слабость лихорадка не имеет специфических характеристик снижение массы тела Они отражают активность патологического процесса, нередко встречаются в дебюте болезни

Клиническая картина СКВ 2. ПОРАЖЕНИЕ КОЖИ, ВОЛОС И НОГТЕЙ: эритему лица в виде «бабочки» подострую кожную красную волчанку дискоидные элементы алопецию панникулит диффузную пальмарную эритему эритему околоногтевых валиков сетчатое ливедо геморрагии и другие

Клиническая картина СКВ эритема лица в виде «бабочки»

Клиническая картина СКВ Фотосенсибилизация - повышенная чувствительность к инсоляции встречается в 45–70% случаев, у половины больных приводит к обострению болезни. Фотоиндуцируемые кожные изменения развиваются на открытых участках тела (лицо, зона «декольте», верхние конечности) и представлены разнообразными макулярными, папулёзными и буллёзными повреждениями, а также классической эритемой, а также в виде изолированных или сливных эритематозных пятен различной величины, резко отграниченных от окружающей здоровой ткани и распространяющихся к периферии -центробежная эритема Биетта

Клиническая картина СКВ Синдром Роуэлла - редко встречающееся поражение кожи в виде многочисленных эритематозных, резко отёчных кольцевидных высыпаний, напоминающих многоформную экссудативную эритему

Клиническая картина СКВ Дискоидная красная волчанка проявляется встречаются у 20–25% больных эритемой инфильтрацией, гиперкератозом и атрофией Излюбленная локализация дискоидных элементов: лицо ушные раковины шея волосистая часть головы реже верхние конечности на слизистой оболочке полости рта с изъязвлением В исходе поражения на месте очагов остаются участки рубцовой атрофии

Клиническая картина СКВ Диссеминированная красная волчанка ещё один вид кожного поражения, образованный множественными, рассеянными по различным участкам кожного покрова очагами дискоидной волчанки Капилляриты отёчная эритема с мелкоточечными геморрагиями на подушечках пальцев рук, ладонях и подошвах Кожные геморрагии у 9–20% больных; как правило, они связаны с тромбоцитопенией или нарушением функции тромбоцитов; могут быть обусловлены приёмом лекарственных препаратов (ГК, НПВП, салицилатов), а также кожным васкулитом. Активный кожный васкулит может быть представлен некротическими язвами конечностей, гангреной пальцев, инфарктами кожи. Хронические язвы нижних конечностей развиваются у 3–5% больных. Вышеперечисленные изменения могут быть проявлениями вторичного АФС.

Клиническая картина СКВ Люпуспанникулит - глубокая красная волчанка Капоши–Ирганга. Редкая форма кожного поражения, проявляется плотными болезненными подкожными инфильтратами на лице, волосистой части головы, конечностях, оставляющими после себя глубокие вдавления. Телеангиэктазии - расширение сосудов без признаков воспаления - неспецифический признак СКВ. Диффузная гиперпигментация - кожи встречается у 5–8% больных СКВ, обычно на открытых участках тела, разгибательных поверхностях конечностей. Возможна депигментация. Алопеция - частый (24–70%), но неспецифический признак СКВ. Поражаются волосистая часть головы, брови, ресницы, подмышечные впадины и другие области. Выделяют несколько форм алопеции: очаговая и диффузная, рубцовая и нерубцовая.

Клиническая картина СКВ Поражение ногтей наблюдают у четверти больных СКВ, чаще при активном процессе. Проявляется диффузной краснотой полулуний, продольной и поперечной исчерченностью, атрофией околоногтевого валика, онихолизисом, лейконихией.

Клиническая картина СКВ ПОРАЖЕНИЕ СЛИЗИСТЫХ ОБОЛОЧЕК: энантема - эритематозные участки с геморрагическими вкраплениями и эрозиями слизистой оболочки. афтозный стоматит - умеренно болезненные язвы. люпусхейлит - белесоватые бляшки неправильных очертаний. Вовлечение в патологический процесс красной каймы губ протекает с выраженным воспалением, отёчностью, трещинами, образованием эрозий и язв, покрытых серозно-кровянистыми корками. Эрозивно-язвенный процесс может сопровождаться жжением и выраженной болезненностью. Исход поражения — атрофия. Красная кайма истончается, на поверхности появляются телеангиэктазии. Длительно существующие изъязвления в полости носа, обусловленные лейкоцитокластическим ангиитом, иногда могут приводить к перфорации носовой перегородки (0,5–1% больных).

Клиническая картина СКВ ПОРАЖЕНИЕ ОПОРНО-ДВИГАТЕЛЬНОГО АППАРАТА Поражение суставов (вовлекаются мелкие суставы кистей, реже поражение затрагивает лучезапястные, локтевые, коленные и другие суставы. Артриты носят симметричный характер, характеризуются рецидивирующим течением. Боль и скованность наблюдаются значительно чаще, чем объективные признаки поражения суставов. Утренняя скованность непродолжительна). Поражение связочного аппарата (тендиниты, тендосиновиты) Поражение скелетной мускулатуры (миалгии, повышенная чувствительность мышц при пальпации, мышечная слабость, атрофия) Асептические некрозы костей (часто развиваются в головке бедренной кости и коленных суставах, реже вовлекаются плечевые, голеностопные и локтевые суставы).

Клиническая картина СКВ ПОРАЖЕНИЕ ДЫХАТЕЛЬНОЙ СИСТЕМЫ: Гортань - неспецифическое воспаление слизистой оболочки гортани, двигательные расстройства (паралич или парез голосовых связок), подглоточный стеноз, воспалительный отёк гортани, различные инфекционные поражения. Плевриты - развиваются чаще на фоне других проявлений СКВ, но встречаются и в дебюте заболевания в виде моноорганного поражения. У большинства больных развитие плеврита сопровождается болью в грудной клетке, одышкой, кашлем, лихорадкой. Выпот чаще двусторонний, объём его небольшой или умеренный, по характеру это экссудат. Лёгочная гипертензия - может быть обусловлена тромбоэмболией ветвей лёгочной артерии, наблюдаемой у 5–12% больных СКВ в рамках вторичного АФС. В большинстве случае признаки лёгочной гипертензии появляются через несколько лет после начала СКВ, развиваются исподволь и имеют тенденцию к постепенному прогрессированию. Острый волчаночный пневмонит Лёгочные (альвеолярные) геморрагии

Клиническая картина СКВ ПОРАЖЕНИЕ СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ: Поражение перикарда характеризуется количеством экссудата небольшое или умеренное. Редкое осложнение перикардита - тампонада сердца, возникающая чаще у молодых больных при активных формах болезни. У части больных развивается гнойный и констриктивный перикардит. Поражение клапанного аппарата – редко. Эндокардит Либмана-Сакса характеризуется поражением одного (чаще митральный) или нескольких клапанов. Страдают преимущественно молодые больные с высокой активностью волчаночного процесса. Формирование стенозов с развитием гемодинамических нарушений встречается редко. Отмечена более высокая, чем в популяции, частота пролапса створок митрального клапана. Патологические процессы в миокарде – чаще протекают в виде миокардита, ГЛЖ, миокардиодистрофии на фоне приема лек-х препаратов. Патология сосудов

Клиническая картина СКВ ПОРАЖЕНИЕ ОРГАНОВ ПИЩЕВАРЕНИЯ: Поражение слизистой оболочки рта проявляются в виде язвенного стоматита, фарингита, диспепсческих явлений и пептических язв. Поражение кишечника связано с вовлечением серозных оболочек и поражением сосудов брыжейки. Васкулит мезентериальных артерий с развитием кишечных инфарктов и изъязвлений относят к наиболее тяжёлым проявлениям. Патология печени Наиболее частые причины желтухи при СКВ (1–4% больных) — гемолитическая анемия и вирусный гепатит, реже обструкция желчевыводящих путей и цирроз печени. Редко развивается васкулит печени, приводящий к инфарктам и спонтанным разрывам органа с картиной острого живота. Асцит основная причина его возникновения - нефротический синдром. Асцит может быть вызван застойной сердечной недостаточностью (при этом отсутствует болевой синдром, а асцитическая жидкость представляет собой транссудат), перитонитом (болевой синдром выражен, определяется экссудат) и циррозом печени (редко).

Клиническая картина СКВ ПОРАЖЕНИЕ ПОЧЕК: У подавляющего большинства развитие волчаночного нефрита наблюдают в течение первых 5 лет болезни, у 3–10% больных поражение почек может быть первым проявлением СКВ. Активные формы волчаночного нефрита развиваются обычно в первые годы заболевания на фоне выраженного иммуновоспалительного процесса, чаще в молодом возрасте. Больным старшего возраста свойственно менее агрессивное течение нефрита (как клинических, так и морфологических вариантов).

Клиническая картина СКВ ПОРАЖЕНИЕ НЕРВНОЙ СИСТЕМЫ: транзиторные ишемические атаки Инсульты Эпилептические припадки Миелопатия Периферические невропатии Вторичные поражения Психические, нервнопсихические и поведенческие проблемы

Клиническая картина СКВ ПОРАЖЕНИЕ ОРГАНОВ ЗРЕНИЯ И СЛУХА: Офтальмологические проявления включают сухой кератоконъюнктивит в рамках вторичного синдрома Шёгрена, эписклерит, иридоциклит, ишемическую нейропатию, неврит зрительного нерва, а также изменения, обусловленные приёмом лекарственных препаратов (задние субкапсулярные катаракты). Вовлечение органа слуха в патологический процесс наблюдают редко (0-3% случаев) в виде среднего отита и неврита слухового нерва с возникновением тугоухости различной степени. ЭНДОКРИННЫЕ НАРУШЕНИЯ: Эндокринные нарушения при СКВ недостаточно изучены, хотя и признается их более высокая распространённость по сравнению с популяцией. Развитие сахарного диабета отмечено у 7–9% больных, что в ряде случаев обусловлено глюкокортикоидной терапией. Частота гипотиреоза составляет 1–10%, гипертиреоза - 3-11%, аутоиммунного тиреоидита Хасимото — 0,5-4%.

ДИАГНОСТИКА СКВ

ДИАГНОСТИКА СКВ

ДИАГНОСТИКА СКВ

Диагностика АНАМНЕСТИЧЕСКИЕ ДАННЫЕ следует обращать внимание на факторы, предшествующие развитию болезни и/или обострений, на динамику клинических и лабораторных показателей в зависимости от менструального цикла у женщин, гинекологический анамнез. Необходимы детальные сведения об аллергических реакциях. Немаловажное значение имеет наследственная предрасположенность (наличие родственников с РЗ, сердечнососудистыми и прочими заболеваниями). Учитывается социально-экономический статус, профессия и условия труда пациентов, регион проживания (север, юг), миграционные данные. ФИЗИКАЛЬНОЕ ОБСЛЕДОВАНИЕ выявляются признаки, отражающие распространённость и активность основного заболевания, наличие сопутствующей патологии, степени тяжести поражения внутренних органов и пр. ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ (ОАК, ОАМ, коагулограмма, биохимия крови, иммунологические исследования) ИНСТРУМЕНТАЛЬНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ (рентгенография костей, суставов, органов грудной клетки, УЗИ суставов и мягких тканей, денситометрия, бронхоскопия, КТ, МРТ, Эхо-КГ, ЭГДС и др.)

Дифференциальная диагностика Проводят с: 1. гематологическими заболеваниями (гемолитическая анемия, идиопатическая тромбоцитопеническая пурпура); 2. системными васкулитами, лимфопролиферативными заболеваниями, другими РЗ (болезнью Шёгрена, первичным АФС, фибромиалгией, ранним РА, ювенильным хроническим артритом); 3. смешанной криоглобулинемией при гепатите C, 4. сывороточной болезнью, 5. синдромом лекарственной волчанки, 6. паранеопластическим синдромом, 7. саркоидозом, воспалительными заболеваниями кишечника, 8. инфекционными заболеваниями (лаймским боррелиозом, туберкулёзом, вторичным сифилисом, инфекционным мононуклеозом, вирусными артритами, ВИЧ-инфекцией, гепатитом B и др.).

Формулировка диагноза Пример: Системная красная волчанка, острое течение, активность 3, поражение почек (люпус-нефрит с нефротическим синдромом и сохранной функцией почек), кожи и слизистых оболочек (фотосенсибилизация, «бабочка», язвенный стоматит, ладонные и подошвенные капилляриты), суставов (мигрирующий полиартрит), серозных оболочек (плеврит, перикардит), системы крови (лейкопения, гемолитическая анемия). Двусторонняя нижнедолевая крупозная пневмония, ДН I.

ЛЕЧЕНИЕ СКВ ЦЕЛЬ ЛЕЧЕНИЯ: Достижение клинико-лабораторной ремиссии заболевания. Предотвращение поражения жизненно важных органов и систем, в первую очередь почек и ЦНС. Улучшение качества жизни. ПОКАЗАНИЯ К ГОСПИТАЛИЗАЦИИ: Лихорадка неясного генеза (инфекционные осложнения — одна из наиболее частых причин смерти больных СКВ). Боли в грудной клетке. Наличие симптомов диффузного поражения ЦНС. Выраженные цитопении. Активные формы волчаночного нефрита и быстропрогрессирующая почечная недостаточность. Острый пневмонит или лёгочное кровотечение. Обострение СКВ (при невозможности коррекции лечения на амбулаторном этапе).

НЕМЕДИКАМЕНТОЗНОЕ ЛЕЧЕНИЕ Общие рекомендации: снижение психоэмоциональной нагрузки, уменьшение пребывания на солнце, активное лечение сопутствующих заболеваний. В период обострения заболевания и на фоне лечении цитотоксическими препаратами показана эффективная контрацепция. Противопоказаны пероральные контрацептивные препараты с высоким содержанием эстрогенов, поскольку они могут вызывать обострение СКВ. Следует избегать введения вакцин и лечебных сывороток. С целью профилактики остеопороза рекомендовано прекращение курения, употребление пищи с высоким содержания кальция, физические упражнения. Для профилактики атеросклероза показана диета с низким содержанием жиров и холестерина, прекращение курения, контроль массы тела, физические упражнения.

МЕДИКАМЕНТОЗНОЕ ЛЕЧЕНИЕ Глюкокортикостероиды: ГК короткого действия (преднизолон и метилпреднизолон) — наиболее эффективные лекарственные средства для лечения СКВ. Доза ГК зависит от активности заболевания: •Больным с низкой активностью заболевания назначают небольшие дозы ГК (преднизолон <10 мг/сут). •При умеренной активности — средние дозы (20–40 мг/сут) в течение 4 нед с постепенным снижением до поддерживающей дозы. •При высокой активности болезни и тяжёлом поражении ЦНС, почек, системы крови (тромбоцитопении, гемолитическая анемия) назначают высокие дозы ГК в сочетании с цитотоксическими препаратами. Абсолютное показание для назначения высоких доз ГК [1 мг/(кг×сут) и более] — быстро прогрессирующее поражение жизненно важных органов! Длительность приёма высоких доз ГК варьирует от 4 до 12 нед в зависимости от выраженности клинического эффекта. Снижение дозы следует проводить постепенно, под тщательным клиниколабораторным контролем, а поддерживающие дозы (5–10 мг/сут) необходимо принимать в течение многих лет. Пульстерапия (500–1000 мг метилпреднизолона внутривенно капельно в течение как минимум 30 мин 3 дня подряд) показана больным с высокой активностью СКВ с целью достижения быстрого эффекта, а также снижения дозы пероральных ГК.

МЕДИКАМЕНТОЗНОЕ ЛЕЧЕНИЕ Цитотоксические препараты: Показания к включению в комплексное лечение больных СКВ цитотоксических препаратов: активный волчаночный нефрит; высокая общая активность болезни; резистентность к ГК, развитие побочных реакций на ГК на ранних этапах лечения (например, явления гиперкортицизма у подростков), необходимость быстрого снижения или уменьшения поддерживающей дозы преднизолона, превышающей 15–20 мг/сут. Циклофосфамид - препарат выбора при волчаночном нефрите и тяжёлом поражении ЦНС. Назначение циклофосфамида ежемесячно по 0,5–1,0 г/м2 внутривенно капельно в течение 6 мес, а затем каждые 3 мес в течение двух лет в сочетании с пульстерапией метилпреднизолоном (по 1,0 г/сут последовательно в течение трёх дней) и пероральным приёмом ГК (преднизолон 40–60 мг/сут) позволяет увеличить выживаемость больных пролиферативным волчаночным нефритом в большей степени, чем монотерапия ГК (в том числе пульстерапия) или комбинирование ГК с азатиоприном. В большинстве случаев применение циклофосфамида позволяет контролировать клинические проявления СКВ, рефрактерные к монотерапии высокими дозами ГК (тромбоцитопения, поражение ЦНС, лёгочные геморрагии, системный васкулит).

МЕДИКАМЕНТОЗНОЕ ЛЕЧЕНИЕ Азатиоприн следует использовать для поддержания индуцированной циклофосфамидом ремиссии волчаночного нефрита, лечения резистентных к ГК форм аутоиммунной гемолитической анемии, тромбоцитопении и торпидных поражений кожи. Комбинированная терапия азатиоприном и ГК способствует повышению общей выживаемости больных волчаночным нефритом. Стандартная терапевтическая доза азатиоприна составляет 2–3 мг/(кг×сут). Максимальный эффект на фоне лечения данным препаратом проявляется не ранее 6-9 мес. Микофенолата мофетил благодаря наличию цитостатической (а не цитотоксической) активности вызывает побочные эффекты реже, чем азатиоприн. Терапевтическая доза составляет 2-3 г/сут и разделяется на 2 приёма с интервалом в 12 ч, поддерживающая доза — 1 г/сут.

МЕДИКАМЕНТОЗНОЕ ЛЕЧЕНИЕ Метотрексат назначается при рефрактерном к монотерапии ГК волчаночном артрите и поражениях кожи. Циклоспорин в дозе <5 мг/(кг×cут) — препарат второго ряда при нефротическом синдроме, связанном с мембранозным волчаночным нефритом, и тромбоцитопении.

МЕДИКАМЕНТОЗНОЕ ЛЕЧЕНИЕ Нестероидные противовоспалительные препараты: НПВП в стандартных терапевтических дозах можно применять для лечения мышечноскелетных проявлений СКВ, лихорадки и умеренно выраженного серозита. У пациентов с вторичным АФС НПВП следует использовать с осторожностью, так как они могут способствовать развитию тромбозов у больных со склонностью к гиперкоагуляции. Аминохинолиновые производные: Гидроксихлорохин используют при поражениях кожи, суставов и конституциональных нарушениях. Его применение позволяет предотвратить развитие обострений СКВ. Кроме того, гидроксихлорохин снижает уровень липидов и уменьшает риск тромботических осложнений. Необходимо проведение полного офтальмологического обследования 1 раз в год в связи с риском развития ретинопатии (1:5000).

Экспертиза нетрудоспособности при СКВ Трудоспособность больных зависит от: характера течения заболевания, степени остроты процесса, тяжести поражения внутренних органов (в первую очередь почек и ЦНС). Определение групп инвалидности проводится в соответствии с общепринятой классификацией и критериями, используемыми при осуществлении медикосоциальной экспертизы.

Диспансерное наблюдение и прогноз Больные подлежат постоянному диспансерному наблюдению с целью выявления признаков обострения заболевания и поражения внутренних органов. В настоящее время прогноз СКВ значительно улучшился: 5летняя выживаемость достигает 86-95%, 10-летняя - 70-80%, 20-летняя - 60-70%. Тем не менее, смертность при СКВ в 3 раза выше, чем в популяции. К факторам неблагоприятного прогноза относят поражение почек (особенно диффузный пролиферативный гломерулонефрит), АГ, мужской пол, начало заболевания в возрасте до 20 лет, сопутствующий АФС, высокую активность заболевания, высокие значения индекса повреждения, присоединение инфекции, осложнения лекарственной терапии.

СИСТЕМНАЯ СКЛЕРОДЕРМИЯ - аутоиммунное заболевание соединительной ткани c характерным поражением кожи, сосудов, опорно-двигательного аппарата и внутренних органов (лёгкие, сердце, пищеварительный тракт, почки), в основе которого лежат нарушения микроциркуляции, воспаление и генерализованный фиброз. ССД относят к группе склеродермических болезней, которая также включает ограниченную (очаговую) склеродермию, диффузный эозинофильный фасциит, склередему Бушке, мультифокальный фиброз, индуцированные формы склеродермии и псевдосклеродермические синдромы.

Эпидемиология ССД Случаи ССД регистрируют во всех регионах мира, однако распространённость заболевания в различных географических зонах и этнических группах неодинакова. Первичная заболеваемость колеблется от 3,7 до 20,0 на 1 млн населения в год. Лица негроидной расы подвержены заболеванию больше, чем представители европеоидной. ССД поражает в основном женщин (соотношение женщин и мужчин — 7:1). Заболевают преимущественно лица 30–50 лет, хотя начало заболевания возможно в любом возрасте (по данным литературы, от 10 мес до 80 лет).

Классификация ССД Клинические формы: • Диффузная форма: - генерализованное поражение кожи конечностей, лица и туловища в течение одного года; синдром Рейно (одновременно или после поражения кожи); - раннее развитие висцеральной патологии (интерстициальное поражение лёгких, поражения ЖКТ, миокарда, почек); - значительная редукция капилляров ногтевого ложа с формированием аваскулярных участков (по данным капилляроскопии ногтевого ложа); - выявление антител к топоизомеразе1 (Scl70).

Классификация ССД • Лимитированная форма: - длительный период изолированного синдрома Рейно; - поражение кожи ограничено областью лица, кистей и стоп; - позднее развитие лёгочной гипертензии, поражения пищеварительного тракта, телеангиэктазий, кальциноза (CRESTсиндром); - расширение капилляров ногтевого ложа без выраженных аваскулярных участков; - выявление антицентромерных антител.
. Характерно сочетание клинических признаков ССД и")
Классификация ССД • Перекрёстная форма (overlap syndrome). Характерно сочетание клинических признаков ССД и ещё одного или нескольких системных заболеваний соединительной ткани. • Висцеральная форма: - отсутствие уплотнения кожи; - синдром Рейно; - признаки лёгочного фиброза, острой склеродермической почки, поражения сердца и ЖКТ; - выявление АНА (Scl70, AЦA).

Классификация ССД • Ювенильная склеродермия: - начало болезни до 16 лет; - поражение кожи нередко по типу очаговой или линейной (гемиформа) склеродермии; склонность к образованию контрактур, возможны аномалии развития конечностей. • Индуцированная склеродермия: распространённое, чаще диффузное поражение кожи (индурация), иногда в сочетании с сосудистой патологией, развившееся после воздействия химических и других факторов внешней среды. • Пресклеродермия: клинически изолированный синдром Рейно в сочетании с капилляроскопическими и/или иммунологическими нарушениями, свойственными ССД.

Классификация ССД Варианты течения: • Острое быстро прогрессирующее течение. Характерно развитие генерализованного фиброза кожи (диффузная форма) и внутренних органов (сердце, лёгкие, почки) в первые два года от начала заболевания. Ранее нередко заканчивалось летальным исходом; современная адекватная терапия улучшила прогноз для этой категории больных. • Подострое умеренно прогрессирующее течение. Характерно преобладание клинических и лабораторных признаков иммунного воспаления (плотный отёк кожи, артрит, миозит), нередки overlapсиндромы. • Хроническое медленно прогрессирующее течение. Характерно преобладание сосудистой патологии: в начале заболевания — многолетний синдром Рейно, постепенно развиваются умеренные кожные изменения (лимитированная форма), нарастает выраженность сосудистых ишемических расстройств, висцеральной патологии (поражение пищеварительного тракта, лёгочная гипертензия).

Классификация ССД Стадии ССД •I -начальная - выявляют 1–3 локализации болезни. •II - стадия генерализации, отражающая системный, полисиндромный характер процесса. •II - поздняя (терминальная), когда имеется недостаточность одного или нескольких органов (сердце, лёгкие, почки). Желательно при формулировке диагноза, определении прогноза и выборе адекватной терапии использовать все три параметра классификации ССД.

Этиология ССД Предполагают, что генез ССД многофакторный, сочетающий наследственную предрасположенность к заболеванию с воздействием неблагоприятных экзо и эндогенных влияний - инфекционных агентов (вирусы и др.), химических веществ, стресса, травмы, охлаждения, эндокринных нарушений и другие.

Патогенез ССД Важнейшее звено патогенеза и морфогенеза ССД - поражение сосудов и нарушение микроциркуляции. Дисфункция эндотелия - наиболее ранний этап патогенеза ССД, лежащий в основе нарушений эндотелийзависимой регуляции кровотока, вазоспастических реакций (синдром Рейно) и последующих структурных сосудистых изменений, приводящих к тяжёлой тканевой ишемии и гипоксии. Характерное для ССД повышенное коллагено и фиброобразование занимает центральное место в патогенезе и определяет нозологическую специфику заболевания.

Клиническая картина ССД ОБЩИЕ ПРОЯВЛЕНИЯ Наиболее характерна значительная потеря массы тела, наблюдаемая в период генерализации или быстрого прогрессирования болезни. Лихорадочная реакция обычно мало выражена (субфебрилитет). Первые проявления ССД - синдром Рейно, поражение кожи (плотный отёк) и суставов (артралгия/артрит).

Клиническая картина ССД Поражение кожи Типичные склеродермические изменения, проходящие стадии плотного отёка, индурации и атрофии, преимущественно локализуются на лице и кистях, нередко сочетаются с сосудистой патологией и трофическими нарушениями (изъязвления, гнойники, деформация ногтей, облысение). Характерна маскообразность лица, первоначально за счёт плотного отёка, а затем индурации и частичной атрофии тканей: отмечаются кисетообразные морщины вокруг рта, уплотнение и натяжение кожи.

Поражение кожи лица и рук

Клиническая картина ССД При ограниченной ССД хронического течения нередки телеангиэктазии, преимущественно на лице, слизистой губ, иногда языка и твёрдого нёба, на груди, спине, конечностях. Склеродактилия - уплотнение (плотный отёк и индурация) кожи кистей с нарастающим ограничением движений и развитием контрактур - характерный признак заболевания, позволяющий, наряду с маскообразностью лица, поставить диагноз уже при первом взгляде на больного.

Cимптом «кисета»

Поражение слизистых, телеангиэктазии

Маскообразное лицо

Клиническая картина ССД Для ранней диагностики необходима ориентация на начальные изменения по типу плотного отёка (особенно изменение пальцев кисти, приобретающих «сосискообразный» вид). При диффузной быстропрогрессирующей ССД развивается практически тотальное поражение кожных покровов, в том числе кожи туловища и конечностей; характерно преобладание индуративных изменений

Поражение кистей

Клиническая картина ССД Выраженность уплотнения кожи оценивают пальпаторно методом «кожного счёта» по 4-балльной системе в 17 анатомических участках (лицо, грудь, живот и симметричные отделы конечностей). Градации: - 0 - отсутствие изменений; - 1 - незначительная плотность кожи (собирается в складку); - 2 - плотность кожи умеренная (собирается в складку с трудом); - 3 - выраженная плотность кожи (доскообразная).

Клиническая картина ССД Сосудистые нарушения Наиболее характерны: - вазомоторные нарушения в виде вазоспастических кризов (синдром Рейно), сопровождающихся побледнением и/или цианозом и чувством онемения пальцев рук, реже ног. Возникают эти расстройства спонтанно или, чаще, при воздействии холода, волнении. Сохраняя, подобно болезни Рейно, преимущественную локализацию в области пальцев рук, синдром Рейно при ССД имеет тенденцию к распространению на кисти и стопы, иногда на подбородок, кончик носа и языка (цианоз и нарушение артикуляции, подобно синдрому «перемежающейся хромоты»).

Язва правой голени Трофические язвы чаще развиваются в области костных выступов, над суставами, иногда в области ушной раковины, лодыжек, пяточной области и т.д.

Оценка выраженности синдрома Рейно

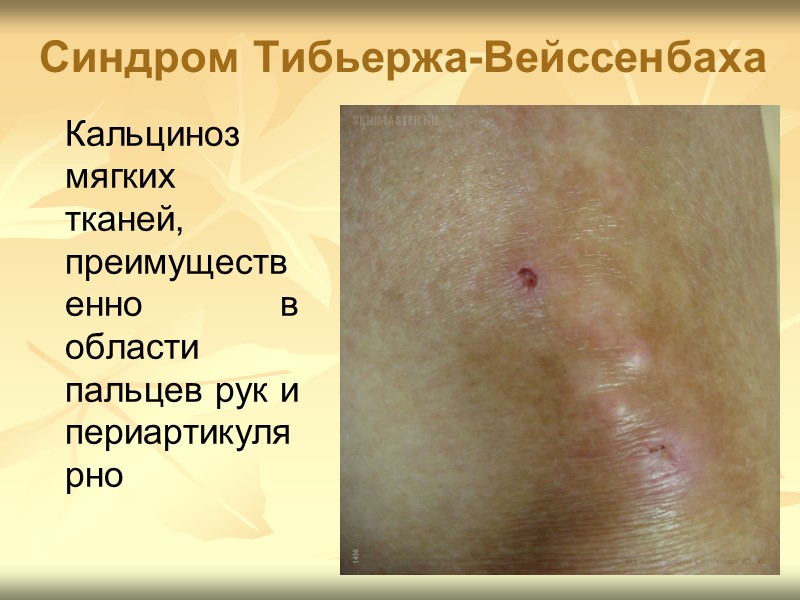
Клиническая картина ССД Поражение опорно-двигательного аппарата Поражение суставов в виде полиартралгий, реже – полиартрита. У части больных развивается ревматоидоподобный полиартрит, но выраженной деструкции суставов не развивается. Поражение скелетных мышц чаще проявляется атрофией и умеренным фиброзом (невоспалительная миопатия), значительно реже — воспалительной миопатией с миалгиями. Возможно поражение костей. Наиболее характерен остеолиз дистальных фаланг, что клинически проявляется характерным укорочением и деформацией пальцев. Реже поражаются отростки лучевых костей и нижней челюсти. Кальциноз мягких тканей, преимущественно в области пальцев рук и периартикулярно (синдром Тибьержа–Вейссенбаха) — часть CRESTсиндрома (сочетание кальциноза, синдрома Рейно, эзофагита, склеродактилии и телеангиэктазии) или лимитированной ССД хронического течения.

Синдром Тибьержа-Вейссенбаха Кальциноз мягких тканей, преимущественно в области пальцев рук и периартикулярно

Клиническая картина ССД Поражение пищеварительного тракта: склеродермический эзофагит эрозии и язвы пищевода гипотония пищевода стриктуры пищевода гипотония желудка синдром мальабсорбции интестинальная псевдообструкция поражения толстой кишки

Клиническая картина ССД Поражение дыхательной системы: Характерны два типа поражения лёгких: • Интерстициальное заболевание лёгких — фиброзирующий альвеолит и диффузный пневмосклероз, преимущественно в базальных отделах лёгких. • Лёгочная гипертензия — изолированная или в сочетании с базальным пневмофиброзом.

Клиническая картина ССД Поражение сердца: Фиброз миокарда желудочков - характерный признак склеродермического поражения сердца, причина систолической и диастолической дисфункции левого желудочка. Аритмии и частые нарушения проводимости: суправентрикулярная тахикардия политопная и групповая экстрасистолия Перикардит (адгезивный, реже экссудативный) обычно протекает бессимптомно Сердечная недостаточность развивается редко, но в случае появления отличается резистентностью к терапии и неблагоприятным прогнозом.
 - быстропрогрессирующая почечная недостаточность,")
Клиническая картина ССД Поражение почек: Острая нефропатия (склеродермический почечный криз) - быстропрогрессирующая почечная недостаточность, злокачественная (гиперрениновая) АГ (однако у 10% больных АД остаётся нормальным), нарастающая протеинурия, олиго и анурия, микроангиопатическая гемолитическая анемия, тромбоцитопения (менее 100 тыс. в 1 мкл), энцефало- и ретинопатия. В настоящее время острая склеродермическая нефропатия развивается реже (возможно, благодаря протективному эффекту современной фармакотерапии). Хроническая латентная нефропатия - протекающей субклинически (преимущественно функциональные нарушения) или с умеренной лабораторной и клинической симптоматикой.

Клиническая картина ССД Поражение нервной и эндокринной системы: Возможны: тригеминальная сенсорная невропатия, чаще в рамках полиневритического синдрома, тиреоидит с развитием гипотиреоза, у отдельных больных - импотенция.

Диагностика ССД АНАМНЕСТИЧЕСКИЕ ДАННЫЕ При опросе больного необходимо обратить внимание на следующие характерные проявления болезни: синдром Рейно, уплотнение кожи, дигитальные язвы, наличие дисфагии и признаков желудочнопищеводного рефлюкса, одышку при обычной для больного нагрузке, боли в груди, перебои в работе сердца, сильные головные боли. Общие симптомы: слабость, быстрая утомляемость, субфебрилитет, потеря массы тела — наблюдаются преимущественно при диффузной ССД, чаще в дебюте болезни. Необходимо уточнить семейный анамнез и предшествовавшие инфекции, травмы, стрессы, воздействие химических веществ и др. ФИЗИКАЛЬНОЕ ОБСЛЕДОВАНИЕ Физикальное обследование позволяет уточнить наличие, характер и степень выраженности основных клинических проявлений ССД и включает оценку следующих параметров: масса тела, выраженность уплотнения кожи («кожный счёт»), наличие синдрома Рейно, дигитальных рубчиков, изъязвлений, гангрены, артрита, теносиновита, шума трения сухожилий, карпального синдрома, уменьшение дистанции «III палец–ладонь», наличие и локализация контрактур, наличие проксимальной мышечной слабости, дисфагии, хрипов в лёгких, шума трения перикарда, нарушений ритма, АГ. Изучение статуса больного в сочетании с лабораторными и инструментальными исследованиями позволяет не только диагностировать ССД, но и выявить особенности клинической картины и течения заболевания, что определяет дальнейшую терапевтическую тактику. ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ (ОАК, ОАМ, коагулограмма, биохимия крови, иммунологические исследования) ИНСТРУМЕНТАЛЬНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ (рентгенография костей, суставов, органов грудной клетки, УЗИ суставов и мягких тканей, капилляроскопия ногтевого ложа, КТ, МРТ, Эхо-КГ, ЭГДС и рН-метрия и др.)
 и линейное (гемиформа по типу")
Дифференциальная диагностика CCД • Ограниченная склеродермия - очаговое (бляшечное) и линейное (гемиформа по типу «удара саблей») поражение кожи и подлежащих тканей. • Диффузный эозинофильный фасциит - индурация тканей начинается с предплечий и/или голеней с возможным распространением на проксимальные отделы конечностей и туловище; пальцы кистей и лицо остаются интактными. Характерны поражение кожи по типу «апельсиновой корки», сгибательные контрактуры, эозинофилия, гипергаммаглобулинемия и повышение СОЭ. Примерно в 1/3 случаев прослеживается связь с предшествовавшей чрезмерной физической нагрузкой или травмой. Возможно развитие апластической анемии. • Склередема Бушке - выраженная индурация в области лица, шеи, плечевого пояса. Часто связана с предшествовавшей инфекцией верхних дыхательных путей. • Мультифокальный фиброз • Опухоль ассоциированная (паранеопластическая) склеродермия

Дифференциальная диагностика CCД • Псевдосклеродермия - изменения кожи, наблюдаемые при врождённых или приобретённых нарушениях метаболизма: порфирия, фенилкетонурия, амилоидоз, синдромы Вернера и Ротмунда; диабетическая псевдосклеродермия, склеромикседема и др. - Порфирия, фенилкетонурия, гликогенозы и мукополисахаридоз могут сопровождаться развитием склеродермоподобного поражения кожи, подлежащих тканей, мышц, стойкими контрактурами, однако в отличие от ССД отсутствуют синдром Рейно, характерная висцеральная патология, иммунные нарушения, а при биопсии выявляют морфологические особенности, свойственные каждой из нозологий. Синдром Вернера (прогерия взрослых, дефект гена ламина) проявляется склеродермоподобными изменениями кожи (особенно конечностей) и скелетных мышц, развитием катаракты, гипогенитализма, преждевременного артериосклероза, инсулярной недостаточности, повышенным риском развития остеосаркомы. Наблюдают чаще у мужчин в возрасте 20-30 лет. Синдром Ротмунда-Томсона (атрофическая пойкилодермия). Характерны пойкилодермия лица и конечностей, двусторонняя катаракта, дистрофия волос, ногтей и зубов, гипогонадизм, нарушения эндохондрального окостенения, артериосклероз и карликовость, гиперпигментация кожи, телеангиэктазии, атрофический дерматоз, анемия; повышение риска развития остеогенной саркомы. Синоним — дистрофия Ротмунда.

Дифференциальная диагностика CCД • Индуцированная склеродермия - может возникнуть под воздействием химических агентов, отдельных лекарств, пищевых добавок и др. Возможны кожная, сосудистая и висцеральная патология, неотличимые от ССД (в связи с этим индуцированная склеродермия включена в классификацию). • Изолированный синдром Рейно - определяет необходимость дифференциальной диагностики ССД с другими системными заболеваниями соединительной ткани: СЗСТ, антисинтетазным синдромом в рамках полимиозита и дерматомиозита. При дифференциальной диагностике вторичного и первичного синдрома Рейно следует учитывать, что последний чаще развивается в молодом возрасте, проявляется преимущественно распространёнными цианозом и гиперемией, нередко сочетается с гипергидрозом (вегетативная дисфункция) и обычно не сопровождается развитием ишемических осложнений.

Формулировка диагноза Пример: Системная склеродермия, диффузная форма, быстропрогрессирующее течение. Плотный отёк/индурация кожи (указать локализацию), синдром Рейно, фиброзирующий альвеолит, поражение миокарда с нарушениями ритма и проводимости.

ЛЕЧЕНИЕ ССД ЦЕЛЬ ЛЕЧЕНИЯ: Коррекция сосудистых нарушений и профилактика осложнений. Замедление прогрессирования фиброза. Профилактика и лечение поражений внутренних органов. Улучшение качества и продолжительности жизни. ПОКАЗАНИЯ К ГОСПИТАЛИЗАЦИИ: Уточнение диагноза и подбор лечения. Впервые выявленная ССД, особенно ранняя стадия диффузной формы. Прогрессирующий синдром Рейно, рецидивирующие язвенные поражения кожи и гангрена пальцев кистей и стоп. Прогрессирующее поражение легких. Появление признаков склеродермического почечного криза. Выраженная анемия.

Немедикаментозное лечение Общие рекомендации: Избегать психоэмоциональных нагрузкок, длительного воздействия холода и вибрации, ношение теплой одежды. Уменьшение пребывания на солнце. Отказ от курения, употребления кофе и напитков, содержащих кофеин. Избегать приема лекарственных средств, вызывающих вазоспазм: симпатомиметики, в-блокаторы. Больным с гипотонией пищевода – дробное частое питание, сон в кровати с приподнятым изголовьем.

Медикаментозное лечение Сосудистая терапия: Существует большой арсенал сосудистых препаратов, из которых на первый план выходят - блокаторы кальциевых каналов (нифедипин и др.), - ингибиторы АПФ (каптоприл и др.) -простагландин E1 (алпростадил, вазапростан). Показания к их назначению: - синдром Рейно и его осложнения (ишемия, некрозы); - лёгочная гипертензия; - почечная АГ.

Медикаментозное лечение Вазодилататоры целесообразно сочетать с антиагрегантами: пентоксифиллином (трентал перорально 400–800 мг/сут, при необходимости внутривенно, или вазонит по 600–1200 мг/сут), дипиридамолом (курантил) (150–200 мг/сут), парентеральным введением реополиглюкина♠ (капельно по 400 мл через день, 8–12 перфузий на курс), другими ангиопротекторами. Целесообразно проведение 2–3 курсов внутривенного введения в год, в интервалах — пероральный приём антиагрегантов. При наличии признаков гиперкоагуляции, микротромбозов рекомендовано включение в терапевтический комплекс антикоагулянтов: гепарина по 5000 ЕД подкожно 2–3 раза в день или надропарин кальция (фраксипарин♠) с последующим переходом на варфарин или фениндион (фенилин♠). Возможно сочетание антикоагулянтной терапии с приёмом антиагрегантов (малые дозы ацетилсалициловой кислоты).

Медикаментозное лечение Противовоспалительная терапия: • НПВП в стандартных терапевтических дозах показаны для лечения мышечносуставных проявлений ССД, стойкой субфебрильной лихорадки (высокая лихорадка для ССД не характерна). • Глюкокортикоиды показаны при прогрессирующем диффузном поражении кожи и клиниколабораторных признаках воспалительной активности (артрит, теносиновит, миозит, серозит) в небольших (15–20 мг/сут) дозах. При фиброзирующем альвеолите доза ГК может быть увеличена до 30–40 мг/сут, при полимиозите (ССД–полимиозит) — до 40–50 мг/сут. Приём высоких доз может увеличить риск развития склеродермического почечного криза. • Циклофосфамид применяют при интерстициальном заболевании лёгких (фиброзирующий альвеолит), ранней ССД, быстропрогрессирующем течении, обычно в комбинации с ГК и пеницилламином. Назначают внутривенно в дозе 800–1000 мг один раз в месяц или внутрь 2 мг/(кг×сут). • Метотрексат показан при выраженном поражении суставов и мышц, перекрёстной форме ССД; способен уменьшить распространённость и выраженность уплотнения кожи, но не влияет на висцеральную патологию. • Аминохинолиновые препараты хлорохин (делагил♠) по 0,25 г/сут или гидроксихлорохин (плаквенил♠) по 0,2–0,4 г/сут нередко включают в комплексную терапию, особенно при хроническом течении ССД.
- антифиброзное действие препарата реализуется при длительном")
Медикаментозное лечение Антифиброзная терапия: Пеницилламин (купренил и др.)- антифиброзное действие препарата реализуется при длительном (не менее 6-12 мес) применении в дозе 450–900 мг в день с последующим снижением и использованием поддерживающих доз (250–300 мг/сут) в течение 2-5 лет. В случае появления побочного действия препарата (дерматит, диспепсические нарушения, нефропатия и др., выявляющиеся у 1/3 больных) необходимы строгий врачебный контроль, прекращение лечения или уменьшение дозы пеницилламина при осложнениях. Наиболее опасны и требуют отмены препарата угнетение кроветворения и нефротоксическое действие.

Хирургическое и экстракорпоральные методы лечения Хирургической лечение применяют редко: это пластические операции (в области лица, кистей и др.), дигитальная симпатэктомия, ампутация, бужирование и эндоскопическая дилатация пищевода при выраженной стриктуре, трансплантация почек. Плазмаферез используют по показаниям в качестве экстракорпорального метода лечения - при активной быстропрогрессирующей ССД, поражении почек, лёгких (в сочетании с патогенетической медикаментозной терапией).

Экспертиза нетрудоспособности при ССД Больные с острой быстро прогрессирующей ССД нетрудоспособны и должны быть переведены на инвалидность. При лимитированной ССД хронического течения больные ограниченно трудоспособны и должны быть освобождены от тяжёлой физической работы, охлаждения, воздействия химических агентов (необходима медикосоциальная экспертиза). Примерные сроки временной нетрудоспособности - 45-90 дней.

Диспансерное наблюдение и прогноз Предвестники неблагоприятного прогноза: диффузная форма; быстропрогрессирующее течение; возраст начала болезни старше 45 лет; мужской пол, фиброз лёгких; лёгочная гипертензия; аритмия; поражение почек в первые 3 года болезни; анемия; высокая СОЭ в первые 1–2 года болезни; протеинурия в начале болезни.

ДЕРМАТОМИОЗИТ - это прогрессирующее системное заболевание, характеризующееся воспалением поперечно-полосатой мускулатуры, кожных покровов и сосудов микроциркуляторного русла с поражением внутренних органов, нередко осложняющееся кальцинозом и гнойной инфекцией.
 • Первичный идиопатический дерматомиозит. • Миозит, ассоциированный с")
Классификация ИВМ (в модификации Miller, 1994) • Первичный идиопатический дерматомиозит. • Миозит, ассоциированный с системными заболеваниями соединительной ткани. • Ювенильный полимиозит/дерматомиозит. • Миозит, сочетающийся со злокачественными опухолями. • Миозит с «включениями». • Другие формы воспалительных миопатий: - гранулематозный миозит; - эозинофильный миозит; - миозит при васкулитах; - орбитальный миозит (глазных мышц); - фокальный (узелковый) миозит; - оссифицирующий миозит.

Этиология дерматомиозита роль генетической предрасположенности указывает возможность развития полимиозита/дерматомиозита у монозиготных близнецов и кровных родственников больных. о возможной этиологической роли инфекционных факторов косвенно свидетельствует более частое начало заболевания зимой и ранней весной (особенно у больных с ювенильным дерматомиозитом), что по времени совпадает с эпидемиями инфекций.

Патогенез дерматомиозита Полимиозит/дерматомиозит рассматривают как аутоиммунные заболевания, связанные с развитием воспалительной клеточной инфильтрации скелетной мышцы и иммунологических нарушений (в частности, с продукцией аутоантител). При иммуногистологическом исследовании поражённых мышц обнаруживают мононуклеарную инфильтрацию Т и Влимфоцитами и макрофагами. Дерматомиозит Полимиозит Преобладают гуморальные Преобладают клеточные иммунные нарушения иммунные нарушения

Клиническая картина ПОРАЖЕНИЯ ОПОРНО-ДВИГАТЕЛЬНОГО АППАРАТА И МЯГКИХ ТКАНЕЙ Поражение мышц Это главный клинический признак заболевания, проявляющийся симметричной слабостью проксимальных групп мышц верхних и нижних конечностей, а также мышц, участвующих в сгибании шеи (поза «кучера»). Характерно поражение мышц: глотки, гортани и верхней трети пищевода, что проявляется дисфонией, дисфагией. Поражение дистальной мускулатуры не характерно, и его отмечают главным образом при миозите с «включениями».

Клиническая картина Поражение кожи характерный признак дерматомиозита ! часто предшествует развитию мышечной слабости. К симптомам поражения кожи относят «гелиотропную» сыпь, папулы Готтрона и симптом Готтрона. и другие кожные проявления: - шелушение и трещины кожи на кончиках пальцев рук («рука механика»); - околоногтевая эритема; - гипертрофия кутикулы; - фотодерматит; - инфаркты околоногтевого ложа; - петехии; - сетчатое ливедо; - телеангиэктазии; - кожный зуд; - панникулит (редко).

Папулы Готтрона

Папулы Готтрона при увеличении

«Гелиотропная» сыпь

Поражение кутикулы и околоногте-вая эритема

Феномен Рейно чаще возникает при дерматомиозите, антисинтетазном синдроме и у больных с перёкрестным синдромом полимиозит/дерматомиозит/ССД. Кальциноз мягких тканей наиболее часто наблюдают при ювенильном дерматомиозите или в поздних стадиях полимиозита/дерматомиозита. Кальцинаты расположены подкожно в соединительной ткани, вокруг мышечных волокон, в зонах микротравматизации над локтевыми и коленными суставами, на сгибательных поверхностях пальцев рук и ягодицах. Поражение суставов при ИВМ не относят к преобладающим клиническим признакам. Возможно преходящее двустороннее симметричное поражение чаще мелких суставов кистей, лучезапястных, реже — локтевых и коленных суставов. В некоторых случаях артриты предшествуют развитию мышечной слабости и напоминают поражение суставов при РА. Отмечают положительный клинический эффект от назначения ГК. Развитие хронического деформирующего артрита с подвывихами суставов кистей наблюдают весьма редко, при этом отсутствуют эрозивные изменения (артропатия Жакку).

Клиническая картина ПОРАЖЕНИЯ ОРГАНОВ ДЫХАНИЯ • Поражение межрёберных мышц и диафрагмы • Пневмония • Фиброзирующий альвеолит (быстропрогрессирующий фиброзирующий альвеолит, более медленное и субклиническое развитие альвеолита)
. Для него")
Фиброзирующий альвеолит Быстропрогрессирующий фиброзирующий альвеолит (синдром Хаммена–Рича). Для него характерны остро возникающий непродуктивный кашель, одышка в покое, лихорадка. При этом симптомы поражения мускулатуры могут уходить на второй план. На рентгенограммах выявляют множественные мелкоочаговые затемнения и сетчатую деформацию лёгочного рисунка. Прогноз неблагоприятный, и такой вариант течения весьма быстро может заканчиваться фатально. Более медленное развитие альвеолита. Заболевание дебютирует одышкой при физической нагрузке, в ряде случаев — непродуктивным кашлем. Эти симптомы могут предшествовать мышечному поражению или возникать на фоне имеющегося тяжёлого миозита. У больных обеих групп аускультативно может присутствовать крепитация в нижних отделах лёгких. В редких случаях наблюдают развитие лёгочного сердца. Симптом «барабанных палочек» возникает редко. Субклиническое поражение. Яркая клиническая лёгочная симптоматика отсутствует. Интерстициальные изменения в лёгочной ткани выявляют при помощи специальных методов исследования (рентгенография, КТ).
. Крайне редко")
Клиническая картина ПОРАЖЕНИЯ СЕРДЦА нарушение ритма и проводимости (тахикардия, аритмия). Крайне редко могут возникать: миокардит, перикардит, застойная сердечная недостаточность.

Клиническая картина ПОРАЖЕНИЯ ЖКТ И ПОЧЕК дисфагия, назальная регургитация жидкой пищи — обусловлены вовлечением в процесс мышц глотки; гипотония пищевода (верхняя треть). Реже возникают рефлюксэзофагит, запор или диарея, ульцерация (особенно при ЮДМ). Почки поражаются крайне редко, как правило, при перекрёстных синдромах, в основном, с ССД. Возможна протеинурия, обусловленная мембранозной нефропатией. Описаны единичные случаи острой почечной недостаточности вследствие массивной миоглобинурии.

Диагностика дерматомиозита АНАМНЕЗ Основные принципы сбора анамнеза у больного с подозрением на полимиозит/дерматомиозит включают выяснение обстоятельств заболевания и ответы на вопросы. Вопросы •Что ограничивает объём движений: боли в мышцах или слабость определённых групп мышц? Каких (проксимальных или дистальных)? •Какие факторы усиливают, а какие уменьшают мышечную слабость? •Приносит ли отдых облегчение? Происходит ли увеличение мышечной силы на фоне отдыха? •За какой промежуток времени произошло нарастание мышечной слабости и как быстро она прогрессирует? Обстоятельства заболевания •Уточняют развитие других (не мышечных) симптомов (утомляемость, сыпь, одышка, нарушение глотания, артралгии). •Определяют связь развития заболевания с воздействием миотоксических веществ (приём препаратов), предшествующей инсоляцией, инфекцией или путешествием. •Определяют симптомы, свидетельствующие о поражении щитовидной железы. •Оценивают вовлечение других мышц (сердца, мышц глотки, ЖКТ, глазных и дыхательных мышц). •Если больной уже получает ГК, выясняют, сколько времени прошло от начала их приёма до нарастания мышечной силы (для ИВМ не характерен быстрый клинический эффект). При сборе анамнеза очень важно отличать мышечную слабость от общей утомляемости или миалгий. Семейный анамнез свидетельствует в пользу наследственной миодистрофии
; б/х крови (увеличение КФК, миоглобина,")
Диагностика дерматомиозита ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ: ОАК (увеличение СОЭ); б/х крови (увеличение КФК, миоглобина, АСаТ и АЛаТ, ЛДГ), иммунологическое исследование (АНФ и аутоантитела – миозитспецифические и миозитассоциированные), Инструментальные: игольчатая электромиография, капилляроскопия, рентгенологическое исследование лёгких, МРТ, холтеровское мониторирование ЭКГ, рентгеновская денситометрия. Обследование, направленное на диагностику онкологических заболеваний, включает определение онкомаркёров: - простатспецифического антигена для диагностики рака предстательной железы; - СА125, СА15.3 для исключение рака яичников и молочной железы и др.

Диагностика дерматомиозита Мышечная биопсия «золотой» стандарт диагностики ИВМ. Её необходимо выполнять даже при наличии характерных клинических, лабораторных и инструментальных признаков заболевания!

Диагностика дерматомиозита Диагностические критерии K. Tanimoto и соавт., 1995: - поражение кожи; - проксимальная мышечная слабость; - увеличение активности КФК и альдолазы; - миалгии при пальпации или спонтанные боли; - первичномышечные изменения при ЭМГ; - антитела Jo1 (гистидилтРНКсинтетаза); - неэрозивный артрит или артралгии; - признаки системного воспаления (лихорадка, СРБ, СОЭ); - морфологические изменения, соответствующие воспалительной миопатии. При наличии первого признака и по крайней мере четырёх из последующих чувствительность для диагностики дерматомиозита составляет 94%, специфичность — 90%; для диагностики полимиозита чувствительность составляет 99%, специфичность — 95%.

Дифференциальная диагностика • Неврогенные заболевания (боковой амиотрофический склероз; спинальная амиотрофия Кугельберга–Веландера; синдром Кеннеди (спинобульбарная мышечная атрофия передних рогов спинного мозга; демиелинизирующие полиневропатии (острая, хроническая); невральная перонеальная амиотрофия Шарко–Мари–Тута. • Первичномышечные заболевания (инфекционные, лекарственные, метаболические, эндокринные, прогрессирующие и непрогрессирующиемышечные дистрофии, рабдомиолиз) • Другие заболевания: ревматическая полимиалгия (выраженные болезненность мышц плечевого и тазового пояса без объективного снижения мышечной силы в сочетании с повышением СОЭ и лихорадкой у лиц старше 50 лет).
, острое (хроническое) течение, слабость проксимальных мышц")
Формулировка диагноза Пример Полимиозит (дерматомиозит), острое (хроническое) течение, слабость проксимальных мышц плечевого и тазового пояса, мышц шеи, дисфони, дисфагия, эритема Готтрона над пястно-фаланговыми, проксимальными межфаланговыми, коленными и локтевыми суставами, артралгии, феномен Рейно, интерстициальный легочной фиброз.

ЛЕЧЕНИЕ ДЕРМАТОМИОЗИТА ЦЕЛИ ЛЕЧЕНИЯ Выделяют несколько целей лечения: - улучшение и сохранение качества жизни; - достижение клиниколабораторной ремиссии; - лечение и профилактика осложнений. ПОКАЗАНИЯ К ГОСПИТАЛИЗАЦИИ Существуют следующие показания к госпитализации больных: - впервые диагностированный полимиозит или дерматомиозит; - обострение заболевания или развитие интеркуррентной инфекции на фоне лечения; - появление симптомов, требующих исключения онкологической патологии.

Немедикаментозное лечение Обучение пациентов проводят по нескольким направлениям: - адаптация к физической активности; - профилактика травматизма (риск переломов вследствие остеопороза); - соблюдение диеты с достаточным содержанием кальция и витамина D (для снижения скорости прогрессирования остеопороза); - строгое исключение продуктов, содержащих углеводы (для снижения риска развития сахарного диабета и ожирения на фоне терапии ГК). Реабилитационные мероприятия необходимо проводить дифференцированно в зависимости от стадии заболевания. В острой фазе показаны пассивные упражнения и напряжение мышц, в стадии выздоровления — изометрические, а затем — изотонические упражнения и, наконец, в хронической стадии — анаэробные упражнения.

Медикаментозное лечение Глюкокортикоиды Принципы лечения ГК: - как можно более раннее начало; - адекватная начальная доза (1,0–2,0 мг/кг в сутки); - длительность приёма начальной дозы не менее 2–3 мес; - медленный темп снижения дозы; - адекватная поддерживающая доза.

Медикаментозное лечение Метотрексат позволяет быстрее перевести больных на поддерживающую дозу ГК. Доза метотрексата варьирует от 7,5 до 25 мг/нед. При плохой переносимости перорального приёма препарата (особенно его высоких доз) целесообразно его вводить парентерально. Азатиоприн в дозе 2–3 мг/кг в сутки (100–200 мг/сут) уступает метотрексату по эффективности и скорости наступления эффекта (в среднем через 6–9 мес), особенно у пациентов с антисинтетазным синдромом. Хлорамбуцил (например, хлорбутин♠). Иногда резистентность к ГК, метотрексату и азатиоприну можно преодолеть, если назначить хлорбутин♠ (2–4 мг/сут) или метотрексат в комбинации с азатиоприном.

Медикаментозное лечение Циклофосфамид редко эффективен при полимиозите/дерматомиозите, но его относят к препаратам выбора при развитии интерстициальных заболеваний лёгких. У больных с резистентным миозитом доза циклофосфамида составляет в среднем 1–2 мг/кг в cутки. Циклоспорин в дозе 2,0–3,5 мг/кг в cутки назначают пациентам при резистентности к ГК. Препарат оказывает положительный эффект при прогрессировании интерстициальных заболеваний лёгких.

Медикаментозное лечение Антималярийные препараты (например, гидроксихлорохин в дозе 200–400 мг/сут) иногда позволяют контролировать кожные проявления дерматомиозита. Их также назначают для поддерживающей терапии в сочетании с низкими дозами ГК. Микофенолята мофетил селективный иммунодепрессант антиметаболического типа действия. Начальная доза составляет 500 мг 2 раза в сутки с последующим её увеличением до 1000 мг 2 раза в сутки. Микофенолата мофетил рекомендуют применять также при резистентном кожном синдроме.

Медикаментозное лечение Плазмаферез следует проводить главным образом больным с тяжёлым, резистентным к другим методам лечения полимиозитом/дерматомиозитом в сочетании с ГК и цитотоксическими препаратами. Внутривенный иммуноглобулин Получены данные о целесообразности внутривенного назначения иммуноглобулина в низких дозах (1 г/кг) два раза в месяц в течение 4–6 мес у больных полимиозитом/дерматомиозитом, резистентным к стандартной терапии ГК и цитотоксическим препаратам. У половины пациентов клиническое улучшение сохраняется в течение трёх и более лет после завершения лечения. Другим возможным показанием к внутривенному введению иммуноглобулина считают тяжёлое поражение пищевода.

Медикаментозное лечение Новые аспекты фармакотерапии В последние годы для лечения ИВМ применяют ингибиторы ФНОα, в частности инфликсимаб. Существуют единичные наблюдения успешного использования при дерматомиозите препаратов, блокирующих пролиферацию Влимфоцитов (ритуксимаб).

Экспертиза нетрудоспособности Сроки нетрудоспособности зависят от своевременности диагностики заболевания и адекватности его лечения. При быстрой постановке диагноза и правильно проведённой терапии нетрудоспособность может не превышать одного года, при тяжёлом течении миозита и его поздней диагностике - составлять от одного до двух лет. При неадекватной терапии и развитии фиброза мышечной ткани вероятна стойкая потеря трудоспособности.

Диспансерное наблюдение и прогноз Ведение больных проводят по нескольким направлениям: •Общий осмотр: при каждом визите (но не реже чем один раз в 2–3 мес). •Определение креатинфосфокиназы: каждые 2–3 мес. •Лабораторное обследование для мониторинга токсичности: в зависимости от характера терапии. •Диспансеризация для проведения онкопоиска: не реже одного раза в год.

Диспансерное наблюдение и прогноз К факторам риска неблагоприятного прогноза при полимиозите/дерматомиозите относят: пожилой возраст, поздно поставленный диагноз, неадекватную терапию в начале болезни, тяжёлое течение миозита, антисинтетазный синдром. Прогноз паранеопластического дерматомиозита зависит от своевременности выявления опухоли и эффективности её лечения.
 - это относительно редкое системное заболевание соединительной ткани")
ДИФФУЗНЫЙ ЭОЗИНОФИЛЬНЫЙ ФАСЦИИТ (болезнь Шульмана) - это относительно редкое системное заболевание соединительной ткани с преимущественным инфильтративно-фиброзным поражением фасций конечностей, подкожной клетчатки, дермы и подлежащих мышц, сопровождающееся эозинофилией и гипергаммаглобулинемией.

Эпидемиология ДЭФ Американский ревматолог L. Shulman в 1974 г. впервые описал двух мужчин, у которых после резкой физической нагрузки появился отек, а затем стойкое уплотнение кожи различных участков конечностей, сопровождавшиеся эозинофилией периферической крови и гипергаммаглобулинемией. При глубокой биопсии обнаружено утолщение подкожной фасции, лимфоцитарная и плазматическая инфильтрация ее. Он предложил наименование болезни «диффузный фасциит». ДЭФ развивается преимущественно в возрасте 25-60 лет, хотя возможно возникновение заболевания в детском и пожилом возрасте. Мужчины болеют в 2 раза чаще, чем женщины.

Этиология и патогенез ДЭФ Этиология и патогенез изучены крайне недостаточно. Известны провоцирующие факторы, они похожи на те, которые обнаружены при ССД: - после физического перенапряжения, - подъема и ношения тяжестей, больших спортивных тренировок, - травмы. Имеется определенная генетическая предрасположенность: семейные случаи очаговой или системной склеродермии, РА и других заболеваний.

Клиническая картина ДЭФ неболезненные уплотнения мягких тканей верхних и нижних конечностей с нарушением их двигательной активности, вплоть до развития стойких сгибательных контрактур в различных суставах, преимущественно пальцев рук; появление чувства стягивания кожи в области верхних и (или) нижних конечностей, ощущение набухания и плотности, реже зуда; структура кожи обычно не меняется, ни в местах уплотнений становится натянутой, блестящей, изредка гиперпигментированной с признаками гиперкератоза; характерен симптом «апельсиновой корки» в виде мягких втяжений при максимальном натяжении, т. е. максимальном разгибании конечности (на внутренней поверхности плеч, бедер); непостоянный субфибрилитет;

Клиническая картина ДЭФ Признаками эозинофильного фасциита являются: частое совпадение начала заболевания со значительной физической перегрузкой; болезненность и уплотнение кожи, обычно дистальных участков конечностей; сгибательные контрактуры; отсутствие синдрома Рейно и висцеральной патологии; эозинофилия крови, увеличение уровня сывороточных иммуноглобулинов; утолщение фасции (подкожной и межмышечных) вследствие развития фиброза и периваскулярных инфильтратов, состоящих из лимфоидных, плазматических клеток и гистиоцитов с наличием эозинофилов; отсутствие выраженных изменений кожи и мышц; благоприятное действие кортикостероидной терапии.
!")
Диагностика ДЭФ Ранний диагностический признак – эозинофилия периферическая и тканевая (до 50%)! Могут также наблюдаться: увеличение СОЭ, наличие СРБ, гиперα2- и γ-глобулинемия, повышение содержания в сыворотке крови серомукоида, фибриногена, церулоплазмина, иммуноглобулинов G и М, ЦИК, повышение титра антинуклеарных и ревматоидного факторов. Это отражает воспалительную и иммунологическую активность заболевания.

Диагностика ДЭФ Диагностическое значение имеет биопсия всего комплекса пораженных при ДЭФ тканей: кожи, подкожной клетчатки, фасции, мышцы.

Дифференциальная диагностика Дифференциальную диагностику следует проводить с: Системной склеродермией, Дерматомиозитом, Склередемой Бушке.

Лечение ДЭФ Глюкокортикостероиды (преднизолон в дозе 20-30 мг/сут до снижения активности процесса, затем дозу снижают до поддерживающей 5-10 мг/сут в течение длительного времени); Цитостатики – назначают при неэффективности ГКС (азатиоприн по 150 мг/сут; при фиброзе – Д-пеницилламин до 450-600 мг/сут); Местная противовоспалительная и антифиброзирующая терапия; При неэффективности терапии – гемосорбция; При снижении активности процесса – массаж, ЛФК.

РЕВМАТИЧЕСКАЯ ПОЛИМИАЛГИЯ воспалительное заболевание опорно-двигательного аппарата, развивающееся только во второй половине жизни человека, характеризующееся сильными болями стереотипной локализации (область шеи, плечевой и тазовый пояс), нарушениями движений, значительным повышением лабораторных показателей воспаления, а также наступлением ремиссии при назначении глюкокортикоидов в небольших дозах.

Эпидемиология РП Частота диагностики новых случаев ревматической полимиалгии за год в различных странах колеблется от 4,9 до 11,1 на 100 тыс. всех жителей. Отмечена тенденция к меньшей распространённости заболевания в странах, расположенных ближе к экватору. У людей моложе 50 лет ревматическая полимиалгия не возникает. Пик заболеваемости наблюдают после 60 лет. Примерно в два раза чаще болеют женщины.

Классификация РП Выделяют: «изолированная» ревматическая полимиалгия; ревматическая полимиалгия, сочетающаяся с гигантоклеточным артериитом.

Клиническая картина РП Боли в области плечевого и тазового пояса двусторонние и симметричные, постоянные, усиливаются при движениях. Типична скованность, наиболее выраженная утром после сна или любого длительного периода неподвижности. Ограничения движений в плечевых, тазобедренных суставах, а также в области шеи. Как правило, воспаление развивается не более чем в трёх суставах, симметричность поражения отсутствует. У отдельных больных развивается слабо выраженный синдром карпального канала с типичным симптомом — онемением в кончиках I–IV пальцев кистей, а иногда ладонный фасциит, вызывающий умеренный отёк кисти, формирование сгибательных контрактур пальцев, уплотнение и болезненность ладонной фасции и сухожилий — сгибателей пальцев.

Диагностика РП Развитие ревматической полимиалгии следует заподозрить у пожилого человека (ранее, как правило, не страдавшего ревматическими заболеваниями) с внезапно, без видимой причины возникающими сильными болями в области плечевых, тазобедренных суставов и шеи, сопровождающимися нарушениями движений, а также неспецифическими симптомами (слабость, субфебрилитет, ухудшение аппетита) и значительным повышением лабораторных показателей воспаления (СОЭ и СРБ).

Диагностика РП Признаки ревматической полимиалгии: возраст пациента в начале болезни старше 65 лет; увеличение СОЭ (более 40 мм/ч); двустороннюю боль симметричного характера в области плечевого и тазового пояса; утреннюю скованность продолжительностью более 1 ч; длительность симптомов более 2 нед; нарастание количества и выраженности клинических симптомов в течение 2 нед; депрессию и/или потерю массы тела; быстрый и значительный эффект преднизолона в суточной дозе не более 15 мг в день. Для диагностики ревматической полимиалгии необходимо наличие всех указанных признаков (чувствительность 99%).
")
Диагностика РП Оценка активности ревматической полимиалгии: SDAI PMR = ВАШ пациента (0–10 см) + ВАШ исследователя (0–10 см) + (утренняя скованность (мин) × 0,1) + элевация верхних конечностей (3–0) + (СОЭ (мм/ч) × 0,1) Оценка индекса активности ревматической полимиалгии: низкая <7; средняя - 7-17; высокая >17.
, псориатический артрит и РА,")
Дифференциальная диагностика парапротеинемические гемобластозы (миеломная болезнь и др.), псориатический артрит и РА, полимиозит, системные васкулиты, заболевания мягких тканей опорно-двигательного аппарата, остеомаляция, гиперпаратиреоз, острые инфекции, сопровождающиеся миалгиями.

Лечение РП Глюкокортикостероиды. Начальная доза преднизолона составляет обычно 15 мг/сут, её обязательно распределяют на 2–3 приёма. Если положительный эффект очевиден, но ко 2–3й неделе лечения не наступает полной клиниколабораторной ремиссии заболевания, дозу преднизолона можно увеличить до 20 мг. После развития ремиссии подавляющую дозу преднизолона сохраняют ещё в течение 1 мес, а затем начинают постепенно снижать на 1,25 мг через каждые 7–10 дней, если нет признаков обострения заболевания, до достижения 10 мг/сут, затем по 1 мг/сут каждые 4 нед до полной отмены препарата. В процессе снижения дозы преднизолона следует тщательно наблюдать за динамикой симптомов: контролировать СОЭ каждый месяц в течение первых 3 мес лечения, затем каждые 8–12 нед в течение 12 мес после завершения лечения.

Прогноз при РП Прогноз у преобладающего большинства больных с изолированной ревматической полимиалгией благоприятен (выздоровление). Если не применяют ГК, заболевание обычно принимает хроническое, волнообразное течение. Известны отдельные случаи спонтанного выздоровления (как правило, не ранее чем через 6-12 мес).

РЕЦИДИВИРУЮЩИЙ ПОЛИХОНДРИТ И ПАННИКУЛИТ довольно редкое воспалительное заболевание неизвестной этиологии, поражающее хрящевые структуры, а также сердечно-сосудистую систему и органы зрения.

Этиология и патогенез Этиология не выяснена. В патогенезе важное значение придают: АТ к коллагену II типа, антинейтрофильным антителам (цитоплазматическе, перинуклеарные).

Патоморфология Потеря гликозаминогликанов хрящевым матриксом Инфильтрация хряща лимфоцитами и плазматическими клетками Образование грануляционной ткани Заключительная стадия - фиброз.

Клиническая картина Поражение хрящей Эпизоды болезненности, отёка и покраснения наружного уха Отёк, размягчение тканей, затем седловидная деформация носа Ринорея При поражении хрящевых отделов гортани и трахеи - кровохарканье, охриплость голоса, затруднение дыхания, рецидивирующие пневмонии

Поражение хрящей ушных раковин

Клиническая картина Поражение слизистых оболочек: язвы в полости рта и гениталий Поражение глаз: кератит, конъюнктивит, склерит, эписклерит, увеит Поражение периферических суставов и позвоночника: асимметричный мигрирующий артрит, хронический симметричный артрит, часто - поражение сочленений между хрящевыми и костными частями рёбер Кожный васкулит Мигрирующие болезненные подкожные узелки, напоминающие тромбофлебит или узловатую эритему Ангионевротический отёк Панникулит Сетчатое livedo Эритема Поражение почек: очаговый пролифера-тивный гломерулонефрит, сегментарный некротизирующий гломерулонефрит Поражение ССС: недостаточность аортального клапана ввиду его растяжения, аневризмы крупных артерий, тромбозы Поражение нервной системы: смешанная сенсомоторная невропатия, невропатия черепных нервов (часто - VIII пары с ослаблением слуха, вестибулопатиями) Лихорадка.

Диагностика Диагноз основан на наличии следующих диагностических критериев: Билатеральный хондрит ушных раковин Неэрозивный воспалительный полиартрит Носовой хондрит Офтальмологические проявления (кератит, конъюнктивит, склерит, эписклерит, увеит) Поражение хрящей гортани или трахеи Слуховые или вестибулярные нарушения Характерны гистологические изменения при биопсии хряща Примечание: Для постановки достоверного диагноза необходимо не менее 3 клинических критериев и гистологическое доказательство.

Дифференциальная диагностика Артериит Такаясу Ревматоидный артрит Системная красная волчанка

Лечение медикаментозное НПВС, например индометацин по 100-150 мг/сут, вольтарен по 100-150 мг/сут. Иммуносупрессивная терапия - при лихорадке, полисиндромном течении, отсутствии эффекта монотерапии от НПВС (однако преимущества иммуносупрессивной терапии не доказаны). Глюкокортикоиды (преднизолон по 30-60 мг/сут со снижением дозы по мере достижения клинического эффекта, далее возможна полная отмена либо назначение поддерживающей дозы 10-15 мг/сут). При офтальмологических поражениях внутриглазничное введение. Циклофосфамид или азатиоприн - в случаях, резистентных к лечению глюкокортикоидами.

Лечение хирургическое Протезирование аортальных клапанов Трахеостомия - при отёке гортани Установка поддерживающих каркасов при поражении хрящевых колец трахеи, сопровождающимся её спадением.
 - системное аутоиммунное заболевание")
СИНДРОМ ШЕГРЕНА Первичный синдром Шегрена (болезнь Шегрена) - системное аутоиммунное заболевание неизвестной этиологии, характеризующееся поражением секретирующих эпителиальных желёз, с вовлечением преимущественно слюнных и слёзных желёз (ксеростомия, ксерофтальмия). Синдром Шегрена (вторичный синдром Шегрена) - аналогичное болезни Шегрена поражение слюнных и слезных желез, развивающееся у 5–25% больных с системными заболеваниями соединительной ткани, чаще при РА, у 50–75% больных с хроническими аутоиммунными поражениями печени (хронический активный гепатит, первичный билиарный цирроз), реже при других аутоиммунных заболеваниях.

Классификация синдрома Шегрена В отечественной классификации по характеру начала и дальнейшему течению болезни Шегрена различают: Хроническая форма (преимущественно железистая) Подострая форма (с внежелезистыми проявлениями)

Клинико-морфологическая и функциональная характеристика




Этиология и патогенез синдрома Шегрена Это следствие иммунопатологических реакций на вирусные антигены; Возможно, это участие вирусов, обладающих сиалотропным (цитомегаловирус, вирус Эпштейна-Барр) и лимфотропным действием (ВИЧ, Т-лимфотропный вирус человека) в развитии данного заболевания; Для заболевания характерен аутоиммунный генез!

Клиническая картина Для подострого течения заболевания характерен дебют в виде одностороннего или двустороннего паротита!

Клиническая картина 2 группы клинических проявлений: 1. Связанные с поражением серетирующих эпителиальных структур; 2. Внежелезистые системные проявления;

Клиническая картина Клинические проявления, связанные с поражением серетирующих эпителиальных структур: Сухой конъюнктивит, сухой кератоконъюнктивит Нитчатый кератит Увеличение слёзных желёз Ксеростомия Рецидивирующий сиаладенит Субмаксиллит/паротит Увеличение поднижнечелюстных/околоушных желёз Увеличение околоушных слюнных желёз I/II степени Сухость слизистых оболочек носа, глотки, гортани, трахеи, бронхов, влагалища Сухость кожи Хронический атрофический гастрит Хронический панкреатит Аутоиммунный холангит Поражение канальцевого аппарата почек

Клиническая картина Внежелезистые системные проявления: Артралгии Рецидивирующий неэрозивный артрит Миалгии/миозит Поражение сосудов Гипергаммаглобулинемическая пурпура Криоглобулинемическая пурпура Смешанная пурпура Уртикарные высыпания Тромбоцитопеническая пурпура Язвенно-некротический васкулит Генерализованный васкулит

Клиническая картина Внежелезистые системные проявления: Синдром Рейно Генерализованная лимфаденопатия Спленомегалия Лимфома Поражение лёгких Выпотной серозит Поражение почек Почечный канальцевый ацидоз I/II типа Тубулоинтерстициальный нефрит Гломерулонефрит/нефротический синдром Острая/хроническая почечная недостаточность Поражение периферической/центральной нервной системы Изолированное поражение периферической нервной системы Изолированное поражение ЦНС Изолированное поражение черепных нервов Комбинированное поражение нервной системы Аутоиммунный тиреоидит Гипотиреоз/тиреотоксикоз

Клиническая картина Различные варианты лимфом, выявленные при болезни Шегрена: Диффузная В-крупноклеточная лимфома Диффузная анапластическая крупноклеточная лимфома с В-клеточным фенотипом Диффузная Т-анапластическая крупноклеточная лимфома с ноль-фенотипом MALT-лимфома Лимфома маргинальной зоны селезенки Фолликулярная лимфома Макроглобулинемия Вальденстрема Лимфома из клеток мантийной зоны Плазмоцитома нодальная (экстрамедуллярная) В-хронический лимфолейкоз Лимфоплазмоцитарная лимфома В-клеточная лимфома, неклассифицируемая малой слюнной железы Т-клеточная лимфома малой слюнной железы Периферическая Т-клеточная лимфома,

Диагностика синдрома Шегрена АНАМНЕЗ В поликлинических условиях в ревматологической практике целесообразно учитывать наличие в анамнезе следующих клинико-лабораторных проявлений: - артралгии, реже неэрозивный артрит мелких суставов кистей, утренняя скованность до 30 мин; - рецидивирующий сиаладенит или постепенное увеличение слюнных желёз; - сухость слизистой оболочки полости рта (носоглотки) и быстрое развитие множественного, преимущественно пришеечного кариеса; - рецидивирующий хронический конъюнктивит; - синдром Рейно или рецидивирующая пурпура; - стойкое увеличение СОЭ; - гипергаммаглобулинемия; - высокие титры РФ. ФИЗИКАЛЬНОЕ ИССЛЕДОВАНИЕ

Диагностика синдрома Шегрена Наиболее информативные показатели при болезни Шегрена: увеличение СОЭ, лейкопения, гипергаммаглобулинемия (60–90%), наличие антинуклеарного и ревматоидного фактора, антител к растворимым ядерным антигенам Ro/SSA, La/SSB (60–100%).

Диагностика синдрома Шегрена ИНСТРУМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ Тест Ширмера: уменьшение слезоотделения после стимуляции нашатырным спиртом (<10 мм/5 мин). Окрашивание эпителия конъюнктивы и роговицы бенгальским розовым и флюоресцеином: это позволяет выявить поверхностные эрозии, очаги дистрофии эпителия, уменьшение времени разрыва слёзной плёнки на роговице <10 с при биомикроскопии.
: I. Сухой")
Диагностика синдрома Шегрена КРИТЕРИИ ДИАГНОЗА БОЛЕЗНИ И СИНДРОМА ШЕГРЕНА (ИНСТИТУТ РЕВМАТОЛОГИИ РАМН): I. Сухой конъюнктивит/кератоконъюнктивит: - уменьшение слезоотделения — стимулированный тест Ширмера <10 мм/5 мин; - окрашивание эпителия конъюнктивы/роговицы бенгальским розовым и флюоресцеином +1 и более; - сокращение времени разрыва прекорнеальной слёзной плёнки < 10 с. II. Паренхиматозный сиаладенит: - сиалометрия: снижение стимулированной секреции слюны < 2,5 мл/5 мин; - обнаружение полостей > 1 мм при сиалографии; - очаговодиффузная лимфогистиоцитарная инфильтрация в биоптатах малых слюнных желёз: более 2 фокусов в 4 мм, при просмотре 2 полей зрения в 4 оцениваемых малых железах. III. Лабораторные признаки аутоиммунного заболевания: - положительный РФ (титр 1:80 и более), или - положительный АНФ (титр 1:160 и более), или - обнаружение Ro/Laантиядерных антител.

Дифференциальная диагностика Синдром Шегрена следует дифференцировать от: заболеваний, протекающих со вторичным синдромом Шегрена: РА, системных заболеваний соединительной ткани (СКВ, ССД, полимиозит/дерматомиозит, перекрёстные синдромы). уменьшение секреции слёзных и слюнных желёз может быть следствием многих причин: (сухость полости рта может быть обусловлена - приемом антидепрессантов, депрессией, дегидратацией, системными заболеваниями – саркоидоз, туберкулез и др.; увеличение в объеме слюнных желез может быть вызвана бактериальными и вирусными инфекциями и др.)
 течения, развёрнутая стадия (начальная,")
Формулировка диагноза Пример Болезнь Шегрена, подострого (хронического) течения, развёрнутая стадия (начальная, поздняя) с поражением слюнных желёз (двусторонний рецидивирующий паренхиматозный паротит, увеличение слюнных желёз I–II степени, гипофункция слюнных желез I–III степени), глаз (сухой конъюнктивит, блефароконъюнктивит, кератоконъюнктивит с дистрофией эпителия конъюнктивы I–II степени и роговицы I–III степени, нитчатый или буллезно-нитчатый кератит, гиполакримия I–III степени), суставов (артралгии, рецидивирующий неэрозивный артрит), кожи (синдром Рейно, рецидивирующая пурпура).

Лечение синдрома Шегрена ЦЕЛИ ЛЕЧЕНИЯ Достижение клинико-лабораторной ремиссии заболевания. Предотвращение развития осложнений заболевания (генерализованный язвеннонекротический васкулит, тяжёлые поражения ЦНС и периферической нервной системы, трансформация в лимфопролиферативные заболевания). Улучшение качества жизни больных.

Немедикаментозное лечение следует ограничивать психоэмоциональную нагрузку. необходимо исключать инсоляцию вследствие плохой переносимости и частого развития фотодерматозов; нежелательно длительное пребывание в районах с сухим, жарким климатом. в помещениях целесообразно использовать увлажнители воздуха. исключить большие голосовые и зрительные нагрузки, работу в запылённых условиях и с химическими веществами. больные должны постоянно применять фторсодержащие и противовоспалительные зубные пасты, проводить санацию очагов инфекции, профилактику грибковой инфекции полости рта и влагалища.
 назначают больным")
Медикаментозное лечение Глюкокортикоиды 5 мг в день или 5 мг через день) назначают больным с рецидивирующими сиаладенитами и минимальными системными проявлениями - суставным синдромом и почечным канальцевым ацидозом. Малые дозы ГКС уменьшают частоту рецидивов сиаладенита, сглаживают системные проявления и улучшают качество жизни, но не увеличивают саливацию. Иммуносупрессивные средства с антилимфопролиферативным действием (хлорамбуцил и циклофосфамид - предпочтительный препарат для лечения васкулита (криоглобулинемический нефрит, поражение ЦНС и периферической нервной системы, язвенно-некротическое поражение кожи). Его назначают в дозе200 мг/нед в течение 2–3 мес с последующим переходом на 400 мг/мес в комбинации с преднизолоном при развитии системных проявлений, угрожающих жизни.

Медикаментозное лечение Пульстерапия и показания к ее проведению: - выпотной серозит; - тяжёлые лекарственные аллергические реакции. Показания к комбинированной пульстерапии: - длительно существующее массивное увеличение слюнных желёз с диффузной инфильтрацией (>400 клеток в поле зрения) и синтезом моноклональных иммуноглобулинов; - ангиоиммунобластная лимфома, MALTлимфома, лимфоплазмоцитарная лимфома без признаков поражения костного мозга; - фокусы лимфоидной инфильтрации в лёгких, альвеолярный лёгочный фиброз; - интерстициальный нефрит, гломерулонефрит с нефротическим синдромом; - язвенно-некротический васкулит; - мононеврит, полиневрит, энцефаломиелополирадикулоневрит, поперечный и восходящий миелит, цереброваскулит; - аутоиммунная гемолитическая анемия, тромбоцитопения.

Прогноз при болезни Шегрена Независимые факторы риска летального исхода: генерализованная лимфаденопатия, спленомегалия, криоглобулинемическая пурпура, полиневропатия, значительное увеличение околоушных желез, анемия, тромбоцитопения, лейкопения, криоглобулинемия, снижение С3/С4фракций комплемента.

СМЕШАННОЕ ЗАБОЛЕВАНИЕ СОЕДИНИТНЬНОЙ ТКАНИ И ПЕРЕКРЕСТНЫЕ ФОРМЫ СИСТЕМНЫХ ЗАБОЛЕВАНИЙ СОЕДИНИТЕЛЬНОЙ ТКАНИ Эти заболевания можно подразделить на три группы: смешанное заболевание соединительной ткани; перекрестные формы системных заболеваний соединительной ткани; недифференцированные заболевания соединительной ткани.

СИНДРОМ ШАРПА - это аутоиммунное заболевание, характеризующееся наличием отдельных признаков системной красной волчанки, системной склеродермии, ревматоидного артрита и дерматомиозита/полимиозита в сочетании с высоким титром антител к экстрагируемому ядерному антигену U1RNP.
,")
Этиология и патогенез Среди этиологических факторов предполагаются: роль инфекции (ретровирусы и др.), генетической предрасположенности (ассоциация с HLADR4), участие механизмов молекулярной мимикрии. Аутоиммунный патогенез заболевания с ведущей ролью нарушений Т-клеточного иммунитета аргументируется наличием иммунопролиферативных процессов в стенках сосудов (васкулиты) и поражённых тканях, а также продукцией аутоантител и иммунных комплексов.

Клиническая картина Начало заболевания характеризуют следующие симптомы: умеренно выраженный синдром Рейно; отек кистей и/или пальцев (по типу «сосисок»); боль в суставах и мышцах, иногда артриты; субфебрилитет. В последующем развивается полиморфная клиническая картина, включающая: синдром Рейно; поражения кожи, суставов, мышц, легких, ЖКТ; поражения сердца, почек и ЦНС, встречающиеся реже.

Диагностика синдрома Шарпа Характерный иммунологический признак, входящий в современные критерии заболевания: высокий титр антиU1РНП-антител, которые могут иметь, как полагают, и прогностическое значение! Диагностический титр – 1:1600 и выше Клинические: Отёк кистей Синовит Миозит Синдром Рейно Акросклероз Требования для постановки диагноза: серологический критерий плюс три клинических (при наличии отёка кистей, синдрома Рейно и акросклероза необходим четвёртый клинический критерий)

Формулировка диагноза Пример Смешанное заболевание соединительной ткани: синдром Рейно, отёк кистей, миалгии, артралгии, антиU1RNP (титр).

Лечение ЦЕЛИ ЛЕЧЕНИЯ Снижение активности. Достижение клинико-лабораторной ремиссии. Сохранение качества жизни. ПОКАЗАНИЯ К ГОСПИТАЛИЗАЦИИ Уточнение диагноза и подбор адекватной терапии. Легочная гипертензия. Поражение легких, почек.
, При необходимости - небольшие")
Медикаментозное лечение Нестероидные противовоспалительные препараты, Глюкокортикостероиды (малые дозы), При необходимости - небольшие дозы метотрексата, аминохинолиновые препараты. При наличии синдрома Рейно показана сосудистая терапия: - блокаторы кальциевых каналов; - пентоксифиллин; - курантил (дипиридамол) и др.
 характеризуются наличием диагностических критериев двух")
ПЕРЕКРЕСНЫЕ ФОРМЫ СИСТЕМНЫХ ЗАБОЛЕВАНИЙ СОЕДИНИТЕЛЬНОЙ ТКАНИ (overlap syndromes) характеризуются наличием диагностических критериев двух и более системных заболеваний соединительной ткани у одного больного.
; Системная склеродермия")
Наиболее частые сочетания: Системная склеродермия и полимиозит (ССДполимиозит или полимиозитССД); Системная склеродермия и ревматиидный артрит (ССДРА); Ревматоидный артрит и системная красная волчанка (РАСКВ); Системная красная волчанка и полимиозит (СКВполимиозит); Ревматоидный артрит и полимиозит (РАполимиозит); Ревматоидный артрит и васкулит (РАваскулит).
29-diffuznye_bolezni_soed_tk.ppt
- Количество слайдов: 187